
The use of alternatives to testing on animals
for the REACH Regulation
1
The use of alternatives
to testing on
animals for the
REACH Regulation
Fourth report under
Article 117(3) of the
REACH Regulation
June 2020

2
The use of alternatives to testing on animals
for the REACH Regulation
LEGAL NOTICE
This publication is solely intended for information purposes and does not necessarily
represent the official opinion of the European Chemicals Agency. The European
Chemicals Agency is not responsible for the use that may be made of the information
contained in this document.
The use of alternatives to testing on animals
for the REACH Regulation
Fourth report (2020) under Article 117(3) of the REACH Regulation
Reference:
ECHA-20-R-08-EN
Cat.
Number: ED-BK-20-001-EN-N
I
SBN: 978-92-9481-734-1
ISSN
: 2600-2590
DOI:
10.2823/092305
Publ. date:
June 2020
Language:
EN
©
European Chemicals Agency, 2020
Cover page © European Chemicals Agency
Reproduction is authorised provided the source is fully acknowledged in the form
“Source: European Chemicals Agency, http://echa.europa.eu/”, and provided written
notification is given
to the ECHA Communication Unit (publications@echa.europa.eu).
If you have questions or comments in relation to this document please send them (quote
the reference and issue date) using the information request form. The information
request form can be acce
ssed via the Contact ECHA page at:
http://echa.europa.eu/about/contact_en.asp
European Chemicals Agency
Mailing address: P.O. Box 400, FI
-00121 Helsinki, Finland
Visiting address: Telakkakatu 6, 00150 Helsinki, Finland
Version
Changes
Date
Version 2.1
Corrigendum:
-
correction of reported statistics
regarding
Skin corrosion/irritation and
Serious eye damage, eye irritation.
- editorial improvement (typos)
09/11/2020
Updated figures:
Figures 1, 2, 3, 13 and 14.
09/11/2020
Updated text in pages:
8, 20, 34, 38-41, 50
Table 6
09/11/2020

The use of alternatives to testing on animals
for the REACH Regulation
3
Contents
Contents ............................................................................................................. 3
Foreword by the Executive Director ................................................................... 5
Summary ............................................................................................................ 7
1 Introduction ............................................................................................... 10
2 LEGAL INSTRUMENTS TO AVOID UNNECESSARY TESTING .......................... 13
2.1 Data sharing and joint submission .............................................................. 13
2.2 Adaptation possibilities of REACH ............................................................... 14
2.3 Testing proposals and third party consultations ............................................ 14
3 Analysis of REACH registrations ................................................................. 16
3.1 Scope: a complete view ............................................................................ 16
3.2 Method ................................................................................................... 17
3.3 Results and discussion .............................................................................. 17
3.3.1 Availability of experimental studies ....................................................... 17
3.3.2 Options used to fulfil requirements in 2016 compared to 2019 ................. 21
3.3.3 Overall trends in the use of alternative methods with an emphasis on higher
tier endpoints ................................................................................................ 26
3.3.4 A more complete view on options used for lower tonnage substances ....... 31
3.3.5 A complete, detailed view per endpoint ................................................. 34
3.3.6 When were studies conducted?............................................................. 36
3.3.7 In-depth analysis: Skin corrosion/irritation and serious eye damage/eye
irritation ....................................................................................................... 39
3.3.8 In-depth analysis: Skin sensitisation ..................................................... 39
3.4 Conclusions from the data analysis ............................................................. 40
4 Robustness of the used adaptations ........................................................... 42
4.1 Testing strategies and adaptations ............................................................. 42
4.2 Use of read-across ................................................................................... 42
4.1 Weight of evidence and data waiving .......................................................... 43
4.2 Use of QSARs .......................................................................................... 43

4
The use of alternatives to testing on animals
for the REACH Regulation
5 Promotion of non-animal test methods....................................................... 45
5.1 Building the knowledgebase of chemicals .................................................... 45
5.1.1 Further work needed by registrants ...................................................... 45
5.1.2 Maximising the availability and use of data ............................................ 46
5.1.3 Using the REACH data for alternatives development: an example ............. 47
5.2 ECHA activities to promote the development of suitable alternatives ............... 47
5.3 Prospects for scientific development ........................................................... 48
6 Conclusions ................................................................................................ 50
Annex 1 ............................................................................................................ 52
A1.1 Description of dossier and substance selection .............................................. 52
A1.2 Processing of endpoint study records ........................................................... 53
A1.3 Aggregation of study information at substance level ...................................... 62
Annex 2 ............................................................................................................ 65
A2.1 Detailed overviews of options used for each endpoint, covering all tonnage bands
...................................................................................................................... 65
Annex 3 ............................................................................................................ 74
A3.1 Detailed results of the options analysis ........................................................ 74

The use of alternatives to testing on animals
for the REACH Regulation
5
Foreword by the Executive Director
Dear reader,
This is now the fourth time that we are presenting our findings to the European
Commission on how companies are using alternatives to testing on animals under
REACH.
One of the main fundamentals of REACH is to strike a balance between gathering
information on possible hazards of chemicals to protect human health and the
environment, and, at the same time, avoiding unnecessary tests on animals by ensuring
that registrants only conduct them when there is no other choice.
This report serves as our vehicle for documenting the current status on alternative
methods and testing strategies that companies are adopting to avoid animal tests. It
confirms our earlier findings that registrants are successfully sharing data and that they
are making extensive use of the different options at their disposal to avoid testing on
animals.
In particular, we see from the report that many companies are avoiding animal tests by
using information on similar substances through read-across. But there is also evidence
that they are providing valid justifications for omitting data, combining evidence from
different sources using weight-of-evidence approaches, predicting properties using
computer models and adopting in vitro methods to isolate tissues, organs or cells rather
than testing on living organisms.
With the completion of the registration deadline back in 2018, companies have
effectively laid their cards on the table. We now have ample data which gives us the
opportunity to comprehensively review how companies have avoided animal tests across
all tonnage bands.
There remains a concerning number of incompliances in registration dossiers with many
still needing to be updated, either voluntarily or after we have requested for this through
a compliance check. We are proactively following up with companies to make sure they
understand that it is their responsibility to provide information that shows their
chemicals can be used safely. I urge companies to take advantage of the guidance,
practical guides, webinars and advice available in our other publications, such as those
on the progress we have made in evaluation, and to use these resources to strengthen
their alternative approaches and avoid unnecessary animal testing.
Chemicals are – and are increasingly going to be – crucial for the survival of Europe’s
manufacturing industry, so having accurate information available is a must. To be
innovative and move towards circularity, we need precise data and in-depth knowledge
as these will underpin our efforts to ensure companies produce safe chemicals, replace
harmful substances with better alternatives, make materials recyclable, reduce animal
testing, and ultimately safeguard our environment and our health.
Our registration database gives us a unique starting point from which to build up a
chemicals knowledgebase to further develop alternative approaches to animal testing.
Such a knowledgebase will be an integral resource for supporting the goals of the
European Green Deal and the Digital Agenda and reinforcing initiatives such as the
chemicals strategy for sustainability, a toxic-free EU environment and the circular
economy.

6
The use of alternatives to testing on animals
for the REACH Regulation
We plan to continue pursuing non-animal testing methods by following the development
of alternative approaches at OECD-level and grasping opportunities for them to be used
in the regulatory arena. And we are also leading and collaborating in various
international projects that seek to promote collaboration and dialogue on the scientific
and regulatory needs for accepting new approach methodologies into regulatory decision
making.
Adopting these approaches will not only allow us to make better informed decisions, but
will also help to minimise the need for studies on vertebrate animals even further.
Bjorn Hansen,
ECHA Executive Director

The use of alternatives to testing on animals
for the REACH Regulation
7
Summary
Every three years, ECHA submits a report to the European Commission on the
implementation and use of non-animal test methods and testing strategies used to
generate information on intrinsic properties and for risk assessment. This report is the
fourth in this series, covering the operational years from 2007 until 2020. It is published
in accordance with ECHA’s obligations under Article 117(3) of the REACH Regulation. The
previous reports were published in 2011, 2014 and 2017.
1
In 2018, as a result of the final REACH registration deadline, ECHA obtained information
for all remaining substances brought on the EU market in quantities between 1 and 100
tonnes per year. This data has given us a unique opportunity to comprehensively review
the status of the use of alternative methods and testing strategies for industrial
chemicals in the EU.
This report recalls the main legal instruments to avoid unnecessary animal testing and
presents a comprehensive analysis of REACH registrations with an update on the use of
alternative methods. Further discussion on the extent to which adaptations to animal
testing are used, as well as aspects of their quality are also provided.
The report also describes ECHA’s activities to promote the use of alternative methods
and adequate application of adaptation possibilities, and to support registrants in
complying with their legal duties.
Finally, it looks ahead to describe potential development areas and to provide thoughts
on how alternative methods may be used in the future.
The report’s main findings are the following:
REACH legal instruments, which are designed to avoid unnecessary animal testing,
continue to largely work well. The data-sharing and inquiry processes remain among the
most effective tools to reduce animal tests. These provisions ensure that test data is
collected, generated and brought together for each substance in one joint registration
dossier, instead of potentially leading to individual submissions.
The last registration deadline has made publically available the large amount of existing
information previously only available to registrants. This information is now
transparently available to support the safe and sustainable use of substances and
avoiding unnecessary animal testing.
For new and existing registrations, in vitro studies for skin corrosion/irritation, serious
eye damage/eye irritation and skin sensitisation have been clearly taken up since 2016.
The amendment of the REACH annexes has certainly played an important role in
accomplishing this change.
In general, no major changes in the use of adaptations have been observed since
the last report in 2017
2
. Furthermore, the following observations can be made:
- Overall, the most commonly used adaptation is read-across, followed by
data waiving, weight of evidence and quantitative structure–activity relationship
(QSAR) models. Experimental studies were available – on average – in 27.1 % of
cases
(-0.5 % compared to 2016).
- When new studies are needed for repeated dose toxicity and toxicity to
1
https://echa.europa.eu/about-us/the-way-we-work/plans-and-reports?panel=animal-testing-reports#animal-
testing-reports
2
The data for the 2017 report was extracted in 2016, which is the date used throughout the report.

8
The use of alternatives to testing on animals
for the REACH Regulation
reproduction screening, these are increasingly performed using the combined
repeated dose toxicity study with the reproduction/developmental toxicity
screening test (OECD 422). This significantly reduces the number of animals
and costs.
- There has been a moderate increase in the availability of pre-natal
developmental toxicity and (sub)chronic repeated dose studies. Decisions
related to compliance checks and testing proposals in the last three years are
likely to account for this.
In general, Annex VII and VIII dossiers received by the 2018 deadline follow the same
patterns in terms of use of adaptations. Furthermore, the following observations can be
made:
- The newly received Annex VIII dossiers follow a similar pattern as dossiers
in higher tonnage bands, with the exception of acute toxicity where the Annex
VIII dossiers have fewer experimental studies (-2.7 %), but weight of evidence,
QSAR and data waiving have increased.
- Remarkably, at REACH Annex VIII, the percentage of short-term toxicity to fish
studies used to fulfill the information requirement decreased since 2016, showing
an effective use of adaptations for this standard information requirement.
However, a minor increase for long-term aquatic experimental studies has been
observed.
- For newly received Annex VII dossiers, fewer experimental studies and
less read-across are observed, with more weight of evidence, QSAR and
data waiving. For dossiers at this Annex level with the lowest data
requirements, it can be concluded that registrants have used alternative
approaches, even more so than in other tonnage bands.
- Annex VII dossiers that were submitted earlier (before 2016) contain more
additional information on top of the standard minimum requirements than the
once submitted later (2018 deadline). Low tonnage substances also needed to be
registered before June 2010, if they were classified as carcinogenic, mutagenic or
toxic to reproduction, in categories 1 or 2 (CMR Cat 1 and 2). These substances
can be expected to have more information than required according to the tonnage
band, since this information was likely forming the basis for their classification in
the past. Information that is needed to classify a substance as CMR Cat 1 and
2, is typically information that is only required starting from Annex IX. In
contrast, the substances that were registered later and are not classified as CMR
Cat 1 and 2, would not have this ‘extra’ information, as this is not required.
Yet, there are still many incompliances in registration dossiers and many still need to
be updated, either voluntarily or after being requested by an ECHA compliance check
decision. ECHA has communicated about this, and in this report some key findings are
summarised. For this report, a spot check of the compliance of stand-alone QSAR
predictions was additionally done and it shows that a substantial number of
predictions are not adequate.
Registrants still have opportunities to strengthen their alternative approaches,
based on the ECHA guidance and tools, as well as the feedback given in other
publications, for example, the progress made in evaluation, according to Article 54 of
REACH.
Looking towards the future, the now complete REACH registration database constitutes a

The use of alternatives to testing on animals
for the REACH Regulation
9
unique starting point of knowledge that can serve the safe use of chemicals as well as
the further development of alternative approaches to animal testing. ECHA has
developed several initiatives in this direction. Also, stimulated by the emerging global
acceptance of the IUCLID data standard to capture and exchange information, ECHA
foresees the possibility to develop an EU chemicals knowledgebase
3
as the basis to
support the European Green Deal
4
and the Digital Agenda and, in particular, to underpin
initiatives and concepts such as the chemicals strategy for sustainability (a toxic-free
environment)
5
, ‘one substance – one assessment’ and the circular economy.
With the chemicals knowledgebase as one of the resources, ECHA will pursue its
objective of promoting non-animal testing methods by developing and maintaining
tools and guidance to support registrants. It will continue to follow and contribute to the
developments at the OECD and to seize opportunities to translate alternative approaches
into the regulatory arena. ECHA is actively supporting the development of the OECD
QSAR Toolbox
6
, a software tool increasingly used in computational toxicology and
chemical hazard assessment.
ECHA is also exploring ways to exploit new approach methodologies (NAMs) with the
ambition to reinforce their applicability in a regulatory context. In this regard, it is
leading and collaborating in various projects involving NAMs within international
consortia such as the APCRA
7
initiative. These approaches are crucial for high throughput
assessment. They will not only allow for better informed decisions but also minimise the
need for studies on (vertebrate) animals, for the protection of human health and the
environment.
3 This platform is probably a significant building block for the OECD Global Knowledgebase. How this data-
platform relates to the “Feasibility study on a common open platform on chemical safety data” currently
executed by DG Environment is to be seen. https://etendering.ted.europa.eu/cft/cft-
document.html?docId=61946
4
https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal_en
5
https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12264-Chemicals-strategy-for-
sustainability
6
http://www.qsartoolbox.org/
7
https://www.epa.gov/chemical-research/accelerating-pace-chemical-risk-assessment-apcra

10
The use of alternatives to testing on animals
for the REACH Regulation
1 Introduction
European context
The EU legislates animal welfare under EU Directive 2010/63/EU
8
on "the protection of
animals used for scientific purposes". This directive is often cited as one of the most
stringent ethical and welfare standards worldwide
9
and implements the ‘3Rs principle’ in
EU legislation: "Principle of replacement, reduction and refinement" first described by
Russell and Burch in 1959
10
.
In February 2020, the European Commission released its 2019 report on the statistics on
the use of animals for scientific purposes in the Member States of the EU in 2015-2017.
11
The report shows that the number of animals used for regulatory compliance of industrial
chemicals is a small fraction of the total number of laboratory animals. In 2017, 9.39
million animals were used for scientific purposes – 69 % were used in research, while 23
% were used to satisfy legislative requirements, ensuring safety for human health
and/or the environment.
The regulatory uses accounted for 2.18 million animals. The majority of regulatory uses
occurred for medicinal products for humans (61 %) and veterinary medicinal products
(15 %). The proportion for industrial chemicals was about 11 %, which represents
approximately 2.5 % of the total animals used for scientific purposes
12
.
The REACH Regulation
REACH’s
13
primary objective is to ensure that human health and the environment receive
a high level of protection. This aim is also balanced with promoting alternative methods
for assessing substance hazards, and the need to enhance the competitiveness and
innovation of industry. The requirement in the REACH Regulation to use alternative
methods whenever possible is based on EU Directive 2010/63/EU on the protection of
animals used for scientific purposes.
ECHA
ECHA
14
was established for managing the implementation of the REACH and CLP
legislation and, in some cases, carrying out the technical, scientific, and administrative
aspects of REACH. It also has to ensure consistency at EU level with respect to these
activities. ECHA helps companies comply with the legislation, advances the safe use of
chemicals, provides information on chemicals and addresses chemicals of concern.
Scope
Under Article 117(3) of the REACH Regulation, “Every three years, the Agency, in
accordance with the objective of promoting non-animal testing methods, shall submit to
the Commission a report on the status of implementation and use of non-animal test
methods and testing strategies used to generate information on intrinsic properties and
for risk assessment to meet the requirements of this Regulation”. This current report is
the fourth edition, fulfilling that obligation.
8
https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:32010L0063
9
https://www.euroscience.org/news/euroscience-supports-directive-201063eu-on-the-protection-of-animals-
used-for-scientific-purposes/
10
Russell, W.M.S.; Burch, R.L. (1959). The Principles of Humane Experimental Technique. Methuen, London.
ISBN 0-900767-78-2.
11
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52020DC0016&from=EN
12
This report covers all testing for the purposes of REACH, irrespective of where the testing takes place i.e.
within or outside of the European Union.
13
https://echa.europa.eu/regulations/reach/legislation
14
https://echa.europa.eu/about-us

The use of alternatives to testing on animals
for the REACH Regulation
11
REACH registration
A key feature of REACH is the greater level of responsibility placed on companies to
ensure safety. REACH is based on the principle that manufacturers, importers and
downstream users are responsible for ensuring and showing that they manufacture,
place on the market, or use substances that do not adversely affect human health or the
environment.
Therefore, registrants are responsible for generating the necessary information to
properly identify and manage the hazards and risks of substances. Registrants are also
responsible for applying alternative methods to avoid unnecessary animal testing, with
REACH stipulating that animal testing is the last resort (Article 13).
REACH specifies the standard information requirements that must be fulfilled in Annexes
VII to X to REACH. These requirements are in relation to the expected tonnages on the
market. The higher the volume, the more information is needed. These requirements are
minimum requirements as they represent the minimum information needed to protect
human health and the environment, by means of classification and labelling and/or risk
assessment. One generic requirement is that the registration dossier should contain all
relevant and available information, regardless of the standard requirements.
Information on intrinsic properties may also be generated in other ways than by tests, as
long as the conditions for adaptations of the standard testing requirements set out in
Annex XI to REACH are met. To address general requirements for generating information
on intrinsic properties of substances, testing on vertebrate animals must only be
undertaken as a last resort.
Where more information on the intrinsic properties of substances is needed, tests have
to be conducted according to the test methods laid down in a Commission Regulation
15
or in accordance with other international test methods that the Commission or ECHA
recognise as being appropriate.
REACH registrations represent the knowledge that companies have on their chemicals,
including existing data from animal testing, alternatives to animal testing for certain
information requirements, and situations where additional animal tests are needed to
ensure safe use (through testing proposals).
The main focus of this report is the analysis of the registration dossiers, as these should
contain all available and relevant information on chemicals on the European market. In
line with the scope of Article 117(3), the focus of this report is on how the registrants
used the alternative methods which are part of the standard requirements (e.g. in-vitro
testing), and how they made use of the legal possibilities to adapt the standard
information requirements (in particular, alternative methods and data waiving).
IUCLID database
Companies report information on the substances they manufacture or import in a
registration dossier submitted to the Agency. The level of information to be submitted
depends on the substance tonnage and its hazardous properties. The registration dossier
must be in IUCLID
16
format. All non-confidential information is published on ECHA’s
website to view
17
or for download
18
.
For this report, ECHA analysed IUCLID registration dossiers for all four tonnage bands
(1-10 tonnes per year, 10-100 tonnes per year, 100-1 000 tonnes per year, and 1 000
15
http://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:32008R0440
16
International Uniform Chemical Information Database
17
https://echa.europa.eu/information-on-chemicals
18
https://iuclid6.echa.europa.eu/web/iuclid/reach-study-results

12
The use of alternatives to testing on animals
for the REACH Regulation
tonnes per year and above according to Article 10 of REACH) corresponding to each
respective information requirement in REACH Annexes VII-X.
Complete view
This fourth edition is the first report published since the 2018 registration deadline
19
, and
therefore covers all substances manufactured or imported in Europe with a volume of one
tonne per year or higher. With this, the coverage of substances in ECHA’s REACH registration
database is complete. The obligation to update a dossier in case of relevant changes
20
, as well
as the requirement to register new substances, makes that the database gives a complete and
up to date view of the industrial chemicals on the European market.
In addition to the availability of experimental studies and the status of implementation
and use of non-animal test methods, the registration database was further analysed to
answer the following specific questions:
1. What are the most commonly used adaptations?
2. Which options did registrants use to fulfil their information requirements for
lower-volume substances (less than 100 tonnes per year)?
3. What are the most noticeable changes for higher-volume substances (100 tonnes
per year and above) since the previous (third) Article 117(3) report?
4. How did the situation evolve for the endpoints where alternative methods have
been introduced since 2016 as standard information requirements?
For technical reasons, the methodology of data analysis used for the previous reports in
this series were redesigned and adapted to take into account significant modifications
introduced by the IUCLID 6 release
21
in 2016. This makes it difficult to directly compare
the data between this edition and previous editions. However, the algorithms developed
for this fourth Article 117(3) report make it now possible to compare the status of the
IUCLD database at different points in time.
The third Article 117(3) report was based on a snapshot of data from 2016. The
algorithms used for this fourth version were executed to reflect the state of the IUCLID
database at two points in time, namely 31 July 2016 and three years later (i.e. 31 July
2019). It is, therefore, possible to examine the time evolution of testing methods and
use of alternatives since the previous report.
For the purpose of this report, a distinction between low-tier and high-tier endpoints was
made according to the following considerations. Endpoints outlined in REACH Annexes VII and
VIII are considered as low-tier endpoints, while endpoints listed in REACH Annexes IX and X
are considered as high-tier endpoints. For the purpose of this analysis, the 28-day repeated
dose toxicity and screening studies for reproductive/developmental toxicity (included in REACH
Annex VIII) are also considered as high-tier endpoints. These studies are closely related to the
high-tier requirements and are often used (in combination with other evidence) in an attempt
to fulfil the repeated dose toxicity and reproductive/developmental toxicity requirements.
Low-tier endpoints include acute rodent toxicity, skin corrosion/irritation, serious eye
damage/eye irritation, skin sensitisation and short-term toxicity to fish. High-tier human
health endpoints include repeated dose toxicity (all routes, all durations), genetic toxicity
in vivo, developmental toxicity, toxicity to reproduction and carcinogenicity. And, high-
tier environmental endpoints include bioaccumulation, long-term fish toxicity and long-
term toxicity to birds.
19
https://echa.europa.eu/-/21-551-chemicals-on-eu-market-now-registered
20
Article 22 of REACH “Further duties of registrants”
21
https://echa.europa.eu/-/iuclid-6-is-available

The use of alternatives to testing on animals
for the REACH Regulation
13
2 LEGAL INSTRUMENTS TO AVOID UNNECESSARY
TESTING
REACH offers different legal instruments to avoid unnecessary testing and to make sure
that (animal) testing is only undertaken as a last resort. The main instruments are data
sharing, adapting information requirements and testing proposals.
2.1 Data sharing and joint submission
All registrants of the same substance have to share data related to vertebrate animals.
They have to agree on the data for their joint REACH registration. It is a collective
responsibility, which applies equally to all co-registrants. If they cannot reach an
agreement, they can submit a dispute to ECHA, which may give them access to data, if
appropriate. ECHA also provides data if a period of more than 12 years has passed after
its submission. In this case, the data can be re-used freely by others for registration.
Data sharing applies to old experimental studies as well as new studies conducted either
spontaneously by registrants to fulfil an information requirement, in preparing their
registration dossier or updating it, or after receiving a request from ECHA following an
evaluation decision.
There are two possible routes for data sharing: pre-registration and establishment of
substance information exchange forums (SIEFs) for existing (phase-in) substances and
inquiry to ECHA for all other substances. Pre-registration ended on 31 May 2017 for
phase-in substances under certain conditions
22
. After this date, the obligatory inquiry
route is the only way to get in contact with other registrants of the same substance.
New contacts between companies for sharing data have continued since the previous
report. For phase-in substances, the earlier trend of around 14 000 pre-registrations
each year on average continued, with 15 000 pre-registrations in 2016. In 2017, there
seemed to be a rush before the closure of pre-registration with over 580 000 pre-
registrations for getting access to SIEFs, and to fulfil the obligatory data-sharing rules.
The inquiry process facilitates data sharing for all registrants who cannot benefit from
the pre-registration mechanism. Since the closure of pre-registration, all substances
enter the system in this way, which led to a significant rise in the number of inquiries to
around 4 200 inquiries per year, with a peak of 6 104 inquiry dossiers around the 2018
deadline. The vast majority of inquiries are to share data for substances previously
registered, with on average only 200-250 substances per year that are new to the
database.
In anticipation of the 2018 registration deadline, the Commission issued an
Implementing Regulation
23
in 2016 to clarify the data-sharing principles and the
requirement that ECHA must ensure that all registrants of the same substance are part
of the same joint submission, even where a registrant separately submits some
information (opt-out). This prompted the need to revise the Guidance on data-sharing
24
.
ECHA also modified REACH-IT, to prevent submissions outside of existing joint
submissions. This ensures that co-registrants discuss sharing of all relevant data for the
substance and avoid duplicating animal tests following the ‘one substance, one
registration’ (OSOR) principle.
22
Phase-in substances below 100 tonnes per year, within six months after exceeding the one tonne per year
threshold.
23
Commission Implementing Regulation (EU) 2016/9 of 5 January 2016 on joint submission of data and data-
sharing in accordance with Regulation (EC) No 1907/2006 of the European Parliament and of the Council
concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH).
24
https://echa.europa.eu/documents/10162/13631/guidance_on_data_sharing_en.pdf

14
The use of alternatives to testing on animals
for the REACH Regulation
With the improvements in IT systems and processes, the number of parallel new
submissions has been reduced to zero. Especially with the high number of additional
registrations related to the 2018 deadline, this change in process and IT has enforced
more data sharing, and has avoided duplicate data generation, including animal testing.
2.2 Adaptation possibilities of REACH
REACH Annex XI(1) specifies the general rules for adaptation of the standard testing
regime set out in annexes VII to X. It provides different options for deviating from the
standard requirements and for using alternative approaches, provided they are duly
justified and scientifically sound. These options are listed as possible adaptations in
REACH Annex XI(1) and include:
1) use of existing data, including historical human data;
2) use of a weight-of-evidence approach;
3) information generated using quantitative structure activity relationships (QSARs);
4) in vitro test methods; and
5) grouping of substances and read-across.
Adaptations can be used either individually or combined in a weight-of-evidence
approach (for example, use of QSAR and information from read-across in combination
with literature evidence or some properties indicating the possible fate of a substance).
In all cases, the data used must be adequate, reliable and relevant for the particular
endpoints, and must follow the criteria set out in Annex XI.
It is also possible to omit (i.e. waive) the standard information required for an endpoint
by other means than the options listed above. REACH Annex XI provides data-waiving
possibilities when testing is not technically possible (REACH Annex XI(2)) or based on
exposure considerations (for example, where no significant exposure can be shown)
(REACH Annex XI(3)).
In addition, for some endpoints, Column 2 of REACH Annexes VII-X gives specific rules
for other adaptation or data-waiving possibilities (for example, based on considerations
of other hazardous properties).
For the analyses conducted for this report, omitting studies as a result of REACH
Annexes VII-X Column 2 adaptations is not distinguished from omitting studies according
to REACH Annex XI adaptations. As such, both options are marked as “data waiver” in
the presented results.
2.3 Testing proposals and third party consultations
For the purposes of registration under REACH, registrants must not undertake any new
studies involving vertebrate animals required by REACH Annex IX or X before submitting
a testing proposal to ECHA and only after receiving receiving ECHA’s decision requiring
the test to be performed, and under which conditions. When they submit their proposal,
the registrants must show that they have considered alternatives
25
in their IUCLID
dossier.
ECHA organises third party consultations for all testing proposals involving vertebrate
animals, for the endpoints specified in REACH Annexes IX and X. The aim is to ensure
that there is no scientifically valid, existing data that could address the hazard endpoint
covered by the testing proposal. Such information, if it can be used to fill the data gap,
may mean that the proposed testing is no longer required and is sent to the registrant
25
https://echa.europa.eu/view-article/-/journal_content/title/considerations-for-alternative-methods-need-to-
be-included-in-your-testing-proposal

The use of alternatives to testing on animals
for the REACH Regulation
15
together with the draft decision for their consideration. ECHA, in consultation with the
Member States, adopts the decision based on the registrant’s proposal, the information
submitted by third parties and any readily available information identified by ECHA.
Many comments received from third parties are about potential strategies that the
registrant could use, for example, information supporting weight of evidence, references
to open literature and, seldom, potentially relevant studies. However, the registrant may
face challenges to make use of this information. One difficulty is to get reliable and
adequate documentation so that the information can be used for classification and risk
assessment and to establish that the information has adequate and reliable coverage of
the key parameters addressed in the corresponding test method. Another challenge is to
get access to study reports identified by third parties and compensate the data owner.
During the last years, the number of comments received decreased significantly. Before
2015, almost all initiated consultations received third party comments, while between
2017 and 2019, only one-third of initiated consultations
26
received third party
comments. As reported previously, the impact of third party consultations has remained
relatively limited for the reasons outlined above. Nevertheless, there are a limited
number of examples of third party comments that pushed registrants to adapt their
testing strategies.
As of 31 December 2019, there were 1 348 information requests stemming from
adopted testing proposal decisions for endpoints concerning vertebrate animal tests (see
Table 1 below). It is not possible to directly correlate these requests with the number of
animal tests that may result. Such requests may address sequential testing strategies
involving the prior conduct of invertebrate tests or may accept the use of data from tests
conducted with another substance (for example, read-across) as plausible.
The most frequent requests in testing proposal decisions are for information for pre-natal
developmental toxicity studies, repeated dose toxicity 90-day studies and toxicity to
reproduction.
Table 1: Number of requests for tests in adopted decisions with testing
proposals taken since the last report (2017-2019) and cumulative number of all
requests since 2009.
Endpoint
(concerning vertebrate
animals only)
Number of requests in
adopted decisions with
testing proposals adopted
since the last report
(2017-2019)
Total number of
requests in adopted
decisions with testing
proposals (2009-2019)
Bioaccumulation
7
25
Long-term toxicity to fish
21
69
Repeated dose toxicity (90-day, all
routes)
103
462
Mutagenicity/genotoxicity in vivo
35
90
Pre-natal developmental toxicity
157
624
26
https://echa.europa.eu/information-on-chemicals/testing-proposals/previous/outcome

16
The use of alternatives to testing on animals
for the REACH Regulation
Toxicity to reproduction
27
72
78
Total
395
1 348
3 Analysis of REACH registrations
Under REACH, registrants are responsible for collecting and generating the necessary
information, including the application of alternative methods, to properly identify and
manage the hazards and risks. They have to make their data and knowledge on
substances transparent, by submitting an electronic REACH registration dossier to ECHA,
using the IUCLID software.
3.1 Scope: a complete view
With this report, the low tonnage phase-in substances, which were registered by the
2018 deadline are also included, providing a complete overview for substances on the
European market within the scope of REACH.
With the update of the data analysis approach, a new feature was introduced to compare
the situation at different time periods. For this report, the situation of 31 July 2016 (the
date when data was extracted for the third 117(3) report), is compared to the current
situation, using data taken from the complete database on 31 July 2019. Comparing the
results for the two cut-off dates gives an insight into the way information requirements
have been fulfilled in 2019 and 2016.
In total, 98 017 dossiers were analysed for this report (see Table 2). These included all
the latest submissions of the 7 553 and 12 184 substances that had been registered
before 31 July 2016 and 31 July 2019, respectively. The analysis was done on
substances for which at least the full Annex VII information requirements are
applicable
28
. For these substances, all the dossiers that (potentially) contain animal tests
or alternative methods to animal testing were considered.
Table 2: Number of substances for each of the REACH annexes (VII – X) which
define the standard information requirements, for the cut-off dates of 31 July
2016 and 31 July 2019.
REACH Annex 31 July 2016 31 July 2019
VII
2 254
4 884
VIII
903
2 642
IX
2 156
2 331
X
2 240
2 327
Total
7 553
12 184
27
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32015R0282&from=CS
28
Dossiers for strictly controlled intermediates (article 17 and 18) were excluded, as well as some of the NONS
substances which were notified under the previous directive 67/548/EEC. More details in Annex 1

The use of alternatives to testing on animals
for the REACH Regulation
17
3.2 Method
ECHA develops algorithms and uses powerful, dedicated data mining tools to screen and
analyse the submitted dossiers. For this report, ECHA also used its in-house scientific
data analysis platform.
As REACH stipulates that registrants need to provide all available and relevant
information, there is often a multiplicity of data and information available for each
required endpoint. This makes it complex to analyse what type of alternative approach
was used.
Different graphs and projections are presented, where choices are made depending on
the nature of the analysis. In some cases, the focus is on all the available information,
for instance, when analysing trends on the use of guideline studies. For other analyses,
the focus is on how registrants fulfilled their requirements.
Therefore, a certain hierarchy needs to be implemented to keep the output graphs
readable. A more detailed technical description of the data extraction, data processing
and graph explanation can be found in Annex 1.
3.3 Results and discussion
The results and discussion are presented from different angles to answer four main
questions for this analysis.
In addition to the availability of experimental studies and the status of implementation
and use of non-animal test methods, the registration database was further analysed to
answer the following specific questions:
1. What are the most commonly used adaptations?
2. Which options did registrants use to fulfil their information requirements for
lower-volume substances (less than 100 tonnes per year)?
3. What are the most noticeable changes for higher-volume substances (100 tonnes
per year and above) since the previous (third) Article 117(3) report?
4. How did the situation evolve since 2016 for the endpoints where alternative
methods have been introduced as standard information requirements?
The evolvement of the skin corrosion/irritation, serious eye damage/eye irritation and
skin sensitisation endpoints are described in more detail in dedicated sections at the end
of this chapter.
3.3.1 Availability of experimental studies
First, information was compiled on all substances for which guideline
29
studies were
available. A comparison between 2016 and 2019 data has been performed to have an
overview of changes in the availability of experimental studies since the last report. An
intuitive way to obtain this overview is to first look at the percentage of substances for
which registrants have provided at least one guideline study for each information
requirement
30
.
Figures 1 and 2 show the results of the study availability analysis. The rows in both
figures represent the endpoints, and each column represents the REACH Annex to which
the substance belongs. Within each cell, the percentage of substances is shown for which
the information requirement is fulfilled with the standard guideline study. This is colour-
coded: the darker the shade of blue, the more guideline studies were provided, this
means that e.g. for developmental toxicity, 19.5% of substances had a guideline study
29
Guideline/experimental study means, an experimental study according to (one of the) guidelines appropriate
to meet the requirement for the endpoint.
30
The values behind the figures in this chapter can be found in Annex 1.3

18
The use of alternatives to testing on animals
for the REACH Regulation
at Annex IX level, which means that 80.5% of substances used some form of an
adaptation.
The standard information requirements for skin corrosion/irritation and serious eye
damage/eye irritation
31
were updated in the legal text on 31 May 2016, and on 10 May
2017 for skin sensitisation
32
, making non-animal testing the default requirement.
Subsequently, in vitro and in vivo studies have been separated in this analysis.
Figure 3 shows the percentage-point difference between 2019 and 2016 (the 2019
percentage minus the 2016 percentage). An increase in the number means that between
2016 and 2019, a higher percentage of substances have at least one reliable (Klimisch
score 1 or 2, as determined by the registrant) guideline study, while a decrease means
that the percentage of substances with such experimental information was reduced and
registrants have used alternative approaches or waiving to fulfil the information
requirement. In some specific cases (for example, skin sensitisation), the changes also
reflect changes in the standard requirements.
It should be noted that non-vertebrate endpoints related to aquatic toxicity have also
been included in the analysis since REACH foresees the use of integrated strategies,
where invertebrates are also considered, which can ultimately affect the number of
studies performed on vertebrate animals.
31
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32016R0863
32
https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:32017R0706

The use of alternatives to testing on animals
for the REACH Regulation
19
Figure 1: Percentage of substances for which guideline studies were used to fulfil
the standard information requirements for each information requirement (2019)
Figure 2: Percentage of substances for which guideline studies were used to fulfil
the standard information requirements for each information requirement (2016)

20
The use of alternatives to testing on animals
for the REACH Regulation
Figure 3: Difference in the percentage of substances for which guideline studies
were used to fulfil the standard information requirements for each information
requirement (percentage in 2019 minus percentage in 2016
In general, Figure 2 shows that endpoints with the highest percentage of guideline
studies were in 2019 for genetic toxicity in vitro, with a percentage of around 50 % to 60
% depending on tonnage, acute toxicity (~ 46–60 %), short-term toxicity to fish (42–50
%). Skin corrosion/irritation in vivo shows a percentage of about 35 % to 45 % of
studies.
On the other hand, bioaccumulation (vertebrates and invertebrates) (<6 %),
carcinogenicity (1–5 %) and toxicity to reproduction (1–8 %) are endpoints for which
typically relatively few guideline studies were available.
On a high level, no significant changes are observed between 2016 and 2019 except in
four areas.
The first area is skin sensitisation, skin corrosion/irritation and serious eye damage/eye
irritation where a significant shift is visible from in vivo to in vitro approaches between
2016 and 2019. This suggests that the Commission Regulation 2016/863 amending
Annexes VII and VIII to the REACH Regulation in 2016
33
has had an impact in terms of
the way registrants are fulfilling information requirements and has contributed to
avoiding animal testing. A further analysis of these endpoints is presented in Sections
3.3.7 and 3.3.8.
The second area that stands out and is visible in Figure 3, is the moderate increase in
the percentage of pre-natal developmental toxicity and (sub)chronic (90-day/28-day)
repeated dose studies for Annex IX and X substances, which is likely to be related to the
decisions ECHA has taken in compliance checks and testing proposals on these
endpoints.
Thirdly, there is an increase in the propensity of the combined repeated dose toxicity
33
http://data.europa.eu/eli/reg/2016/863/oj

The use of alternatives to testing on animals
for the REACH Regulation
21
with the reproduction/developmental toxicity screening test (OECD 422) at Annex VIII
while at the same time, the percentage of repeated dose toxicity (short-term) and
toxicity to reproduction screening studies has lowered compared to 2016. These studies
do not require a testing proposal and can be done on the initiative of registrants. This
increase seems to indicate that when new data has to be generated to ensure safe use,
registrants favour combined tests instead of separated studies which brings significant
reductions in the number of animals and costs.
Fourthly, it can be observed that the percentage of short-term toxicity to fish studies is
lower in 2019 than in 2016, which indicates that the use of adaptations for this standard
information requirement in REACH Annex VIII has increased. It is noticeable that there is
a significant number of short-term fish studies for Annex VII substances, even if there is
no standard information requirement. In addition, a slight increase in experimental data
for long-term aquatic studies in 2019 is visible, which might be a consequence of
industry initiatives to improve dossiers through testing proposals and compliance check
actions by ECHA.
3.3.2 Options used to fulfil requirements in 2016 compared to 2019
REACH gives many options to fulfil the information requirements, and at the same time,
calls for the use of all relevant and available information. The different options that
REACH registrants have as defined in this analysis, are:
- Experimental: the use of an experimental study according a guideline which is in
line with the information requirement.
- Read-across: the use of a guideline study on a different but similar substance to
read-across the results. This includes category approaches of read-across within
groups of substances.
- QSAR: a mathematical prediction relating one or more quantitative parameters,
which are derived from the chemical structure, to a quantitative measure of a
property or activity.
- Weight of evidence: the use of all available and relevant information which
combined would suffice to allow for a conclusion on hazard and risk assesment,
including classification and labelling, without further studies. In Annex 1 , the
combinations of information that lead to the labelling of weight of evidence are
defined.
- Data waiver: omitting the standard information required for an endpoint either by
means of the general REACH Annex XI adaptations (testing is not technically
possible as defined in REACH Annex XI(2)) or based on considerations of
exposure (REACH Annex XI(3)), or by specific Column 2 adaptations of REACH
annexes VII–X.
- Testing proposal: It should be noted that testing proposals remain only for a
period in the database, as they are processed within set deadlines. The number
represented in the graphs reflects the testing proposals at the moment the
snapshot of the database was taken. For an overview of the number of testing
proposal processed, see Chapter 2.3 Testing proposals and third party
consultations.
- Other: other combinations of information that do not match the above defintions,
e.g. literature data.
- No information: the absence of information, most commonly this reflects that the
endpoint is not required and therefore not provided. Another reason for this
category is that the endpoint is part of the integrated testing strategy, and the
test is not required depending on the outcome of other tests.
A direct comparison of the options to fulfil the information requirements, other than
through experimental (animal) testing, is difficult to make because registrants have the
option to combine approaches. For example, a read-across can be combined with QSAR

22
The use of alternatives to testing on animals
for the REACH Regulation
predictions and literature experimental evidence, where none of these pieces of evidence
is adequate on its own.
The approach taken for the analysis is a combination of applying a hierarchy and taking
(arbitrary) decisions on how to label endpoint data. For instance, if an endpoint has a
reliable guideline study, together with a QSAR prediction, this is counted as a guideline
study. The Klimisch score, as assigned by the registrant, was used as a guide to
distinguish reliable information (Klimisch 1 and 2) from other information. If the
registrant used a combination of pieces of information to cover an endpoint, this was
counted as weight of evidence. The approach is explained in full detail in Annex 1.
The results of the analyses are shown in Figures 4 and 5. They show the options used by
registrants to fulfil the information requirements on 31 July 2016 and on 31 July 2019. In
these two figures, data was aggregated (at IUCLID section level) from all processed
dossiers regardless of the tonnage band. Endpoints (listed on the vertical axis) are blue or
red. Blue represents obligatory endpoints, and the percentage is expressed using only the
substances for which the endpoint is requirement. The red endpoints are not obligatory
which means they are either:
- part of an integrated testing approach, and therefore not always obligatory as
these tests are conditional (relevant for Figures 4-11), or
- not required at the level of the annex, and information is provided on top of the
standard minumum requirements (relevant for Figures 6-11)
For the endpoints listed in red on the Y-axis, all substances in their respective tonnage
bands are used to express the percentages. This explains why for the red endpoints “no
information” is a much larger category than for the blue endpoints, as this represents
optional endpoint information.
A more detailed technical description of the data extraction, data processing and graph
explanation can be found in Annex 1. The numerical results can be found in Annex 3.

The use of alternatives to testing on animals
for the REACH Regulation
23
Figure 4: Frequency of the different options to fulfil the information requirements
in 2019 (aggregated at IUCLID section level).

24
The use of alternatives to testing on animals
for the REACH Regulation
Figure 5: Frequency of the different options to fulfil the information requirements
in 2016 (aggregated at IUCLID section level).
From the results presented in Figures 4 and 5, it can be observed that at the highest
level of aggregation, there are no remarkable differences between the approaches used
to fulfil the information requirements in 2016 and 2019. The overall picture has not
changed despite the fact that many lower tonnage dossiers were added due to the 2018
registration deadline, and at least six years of maintaining the existing dossiers (2010
and 2013 deadline) as reflected in Table 3.

The use of alternatives to testing on animals
for the REACH Regulation
25
Table 3: options used to fulfil the information requirements on average, 2019
compared to 2016
Option used
2019
average [%]
2016
average [%]
Experimental
27.1
27.6
Read-across/category
25.1
27.7
QSAR
2.6
3.0
Weight of evidence
3.7
3.7
Other
4.8
5.6
Data waiver
7.7
10.8
Testing proposal
0.2
0.3
No information
28.7
21.2
There are two areas that are worth highlighting as, even in this highly aggregated
overview, they stand out.
Firstly, as can be seen in Figure 4, waiving is used as the most frequent adaptation for
long-term toxicity to fish, suggesting that information on short-term aquatic toxicity and
long-term toxicity for non-vertebrate species has often been considered sufficient to carry
out a chemical safety assessment.
Data on invertebrates for environmental endpoints have been included in the data
analysis so the impact of testing strategies to avoid vertebrate testing with fish can be
explored. REACH Guidance R.7 stipulates that fish testing can be omitted if the fish is
less sensitive than aquatic invertebrates or algae. While the percentage of short-term
toxicity studies with fish is only slightly lower than the percentage of short-term toxicity
studies with aquatic invertebrates (daphnids) and algae, the percentage of experimental
long-term fish studies is substantially lower than that of long-term studies with aquatic
invertebrates. This suggests that the promoted testing strategy to avoid vertebrate
testing has been widely applied. Similarly to the situation in 2016, bioaccumulation (11.5
%), long-term toxicity to fish (almost 4 %) and short-term toxicity to fish (3.1 %) are
the endpoints that require vertebrate animal testing for which (Q)SARs were used most
frequently to fulfil the information requirements without relying on other information.
Secondly, for a number of endpoints the level of ‘no information’ seems higher in 2019
than in 2016. The higher tier endpoints especially show this: carcinogenicity (11 %
difference), toxicity to reproduction (9 % difference), developmental toxicity (16 %
difference). These results suggest that there was more additional information, beyond
the minimum requirements, in the dossiers submitted earlier. Further detailed analysis
reveals that this is due to the nature of the 2019 dossiers (See Chapter 3.3.4).

26
The use of alternatives to testing on animals
for the REACH Regulation
3.3.3 Overall trends in the use of alternative methods with an emphasis
on higher tier endpoints
The third edition of the Article 117(3) report focused mostly on the higher tonnage
substances (Annex IX and X) as most substances in these tonnage bands were
registered before the deadlines in 2010 (Annex X) and 2013 (Annex IX). So, for the
purposes of this fourth edition of the report we investigated if and how the use of
alternative methods for substances in these tonnage bands has evolved. It is important
to note that the pool of substances for these two tonnage bands has been very stable
between 2016 and 2019: 4327 substances were present both in 2016 and 2019 (Annex
IX and X combined). Only few new substances occurred in these tonnage bands: 331
new substances from 2016 to 2019. Even less substances no longer exist at this tonnage
band: -69 substances from 2016 to 2019. Changes in how registrants used the different
options to meet the requirements for these tonnage bands are therefore expected to be
caused by changes of the existing registrations, e.g. spontaneous updates, testing
proposals, and ECHA’s compliance check decisions.
Figures 6 to 9 provide the results for Annex IX and X dossiers for 2016 and 2019. As in
Figures 4 and 5, endpoints in red font are part of an integrated testing approach and are
not always required as they may also depend on the tonnage band.

The use of alternatives to testing on animals
for the REACH Regulation
27
Figure 6: Frequency of the different options to fulfil the information requirements
for Annex X substances in 2019 (aggregated at IUCLID section level).

28
The use of alternatives to testing on animals
for the REACH Regulation
Figure 7: Frequency of the different options to fulfil the information requirements
for Annex X substances in 2016 (aggregated at IUCLID section level)

The use of alternatives to testing on animals
for the REACH Regulation
29
Figure 8: Frequency of the different options to fulfil the information requirements
for Annex IX substances in 2019 (aggregated at IUCLID section level)

30
The use of alternatives to testing on animals
for the REACH Regulation
Figure 9: Frequency of the different options to fulfil the information requirements
for Annex IX substances in 2016 (aggregated at IUCLID section level)
Figures 6 and 7 show that for Annex X substances there are hardly any changes
detectable between 2016 and 2019. The largest change, with an increase of 3 % for
weight of evidence and a decrease of around 3 % for the use of data waiver, is observed
in bioaccumulation.

The use of alternatives to testing on animals
for the REACH Regulation
31
Developmental toxicity – teratogenicity sees a small increase in experimental studies
(1.2 %), and an increase in read-across (2.8 %), somewhat balanced with fewer testing
proposals (-1.9 %) and less data waiving (-1.2 %).
Skin corrosion – irritation and serious eye damage – eye irritation endpoints see a shift
from weight of evidence (2.2 % less in both endpoints) to read-across (2 % for skin
corrosion – irritation and 1.6 % for serious eye damage – eye irritation). These examples
represent the biggest changes, which indeed confirms that these dossiers have been
stable in how endpoints are addressed.
For Annex IX (Figures 8 and 9), the overall picture is also very similar between 2016 and
2019 but some changes are more clearly visible, especially for higher tier human health
endpoints and long-term toxicity to aquatic invertebrates.
Developmental toxicity – teratogenicity has shifted from testing proposal (5 % lower in
2019) to an increase in experimental studies (7.1 % higher) in the 2016-2019 period.
For repeated dose toxicity, an increase in experimental tests of 3.1 % can be observed
in the period of analysis. There has also been a slight increase in the percentage of
experimental long-term toxicity to fish studies (+1.4 % and +0.3 % for Annex IX and
Annex X, respectively), although the overall percentage of substances with experimental
data for this endpoint remains low (5 % and 7.1 % for Annex IX and X in 2019,
respectively).
Generally speaking, adaptations continue to be used more than experimental studies,
with read-across being the most popular option used. A small shift in experimental
studies for some endpoints is visible, but this doesn’t change the overall picture.
3.3.4 A more complete view on options used for lower tonnage
substances
As this is the first time ECHA has been able to execute the analysis with all the lower
tonnage dossiers (Annex VII and VIII) available, this has allowed us to focus on how
alternative methods have been used for this group of substances.
Figures 10 and 11 provide the results for Annex VIII and VII dossiers for 2019.

32
The use of alternatives to testing on animals
for the REACH Regulation
Figure 10: Frequency of the different options to fulfil the information
requirements for Annex VIII substances in 2019 (aggregated at IUCLID section
level). Endpoints labelled in red font are not part of the standard information
requirements at the given tonnage level, or are part of an integrated approach
and are not always required.

The use of alternatives to testing on animals
for the REACH Regulation
33
Figure 11: Frequency of the different options to fulfil the information
requirements for Annex VII substances in 2019 (aggregated at IUCLID section
level). Endpoints labelled in red font are not part of the standard information
requirements at the given tonnage level, or are part of an integrated approach
and are not always required (genetic toxicity in vivo for this annex).

34
The use of alternatives to testing on animals
for the REACH Regulation
The general distribution of the adaptation options for Annexes VII and VIII follows the
overall pattern observed previously for the higher tier endpoints: read-across is the most
popular, followed by data waiving, weight of evidence and QSARs.
When comparing 2019 Annex VIII dossiers (Figure 10) with 2019 Annex IX (Figure 8) for
the endpoints that are required at both tonnage bands, no significant differences are
observed, with the exception of acute toxicity where Annex VIII has fewer experimental
studies (-3.1 %), which is compensated by an increase in the other options (weight of
evidence, QSAR and data waiver).
When comparing 2019 Annex VII dossiers (Figure 11) with 2019 Annex IX (Figure 8) for
the endpoints that are required at both tonnage bands, a generic trend can be observed
of fewer experimental studies, less read across, balanced with more weight of evidence,
QSAR and data waiver at the lower tonnage band. It can be concluded that for the
dossiers with the lowest data requirements, registrants have used alternative
approaches, even more than in the other tonnage bands.
Under Figure 10, it is remarkable that for 37 % of the substances, some information on
the bioaccumulation endpoint was submitted, even though bioaccumulation at Annex
VIII only needs to be followed up, if screening information indicates a potential
persistent, bioaccumulative and toxic (PBT) concern. As context, for the Annex IX and X
substances this information was present for roughly 50 %. Bioaccumulation does not
need to be assessed for substances that have a low potential for bioaccumulation (for
example, based on a low octanol/water partition coefficients (logK
ow
)) or a low potential
to cross biological membranes (in the case of high molecular weight substances).
When discussing the overall difference between 2019 and 2016 dossiers (see Chapter
3.3.2), it was observed that the level of ‘no information’ seems higher in 2019 than in
2016 for a number of endpoints. This is particularly shown for the higher tier endpoints:
carcinogenicity (11 % difference), toxicity to reproduction (9 % difference) and
developmental toxicity (16 % difference). This significant difference does not occur in
any of the other annexes (Annex X and Annex IX). It is, therefore, the result of
differences in the Annex VII and VIII dossiers received until 2016 and the Annex VII and
VIII dossiers received until 2019, respectively. Low tonnage substances also needed to
be registered before June 2010, if they were classified as carcinogenic, mutagenic or
toxic to reproduction, in categories 1 or 2 (CMR Cat 1 and 2). These substances can be
expected to have more information than required according to the tonnage band, since
this information was likely forming the basis for their classification in the past.
Information that is needed to classify a substance as CMR Cat 1 and 2 is typically
information that is only required starting from Annex IX. In contrast, the substances that
were registered later and are not classified as CMR Cat 1 and 2 would not have this
‘extra’ information, as this is not required.
3.3.5 A complete, detailed view per endpoint
Registrants often submit multiple pieces of evidence to cover an information
requirement. Consequently, the projections discussed earlier only show the main
adaptation option per endpoint and cannot fully represent reality. To illustrate that, a
further analysis of the main types of information submitted per tonnage band was
performed, regardless of whether the information was required or not.
Figure 12 shows how registrants can combine the main ways of fulfilling the information
requirements for one endpoint under REACH, for example, experimental data, read-
across and QSAR. Weight of evidence is not shown separately because it is mostly a
combination of different study result types (experiment, read-across, QSAR or data
waiver). In Figure 12, example a) of one low tier (acute toxicity) and b) of one high tier
endpoint (repeated dose toxicity) is presented, to illustrate the information available.
The corresponding plots for all endpoints analysed can be found in Annex 2.

The use of alternatives to testing on animals
for the REACH Regulation
35
Figure 12: For each annex, the slice shows the options used to fulfil the
information requirement: dark blue = experimental, blue = read
across/category approach, light blue = QSAR. See the text for a detailed
explanation.
a) Acute Toxicity (all routes)
b) Repeated dose toxicity (all routes)

36
The use of alternatives to testing on animals
for the REACH Regulation
Acute toxicity is the lower tier endpoint requiring testing on vertebrate animals for which
there is the highest proportion of experimental data, with more than 50 % of substances
covered by reliable guideline studies (Figure 11). However, for a significant percentage
of substances, experimental studies are often submitted together with read-across or
QSARs. In addition, there is quite a significant proportion of adaptations, which are
combined with other evidence. In general, for approximately one-third of the
substances, acute toxicity is covered by multiple options. If we take, for example, the 2
327 substances covered by Annex X (substances registered above 1 000 tonnes per
year), we see that there are actual test data for 53.6 % of the substances. However, for
21.5 % of the total, there are additional read across/category justifications provided in
the dossiers whereas for another 0.3 % QSARs are also provided. For a very small
fraction, the dossiers contain information using all options. For substances for which no
tests are available, the majority of the justifications are using the read across/category
option.
Repeated dose toxicity is the higher tier endpoint for which most guideline studies are
available. Here, the standalone read-across is the most frequently used option (for over
48 % of substances at Annexes VIII-X, read-across is used to cover this endpoint)
followed by standalone experimental studies (approximately 30 %). Other substances
are covered by multiple options, for example, approximately 30 % use read-across and
experimental studies.
These two examples show that there is a significant proportion of endpoints covered with
multiple options. These alternative options are in fact more abundantly applied than
might appear from the earlier sections, which give a more simplified view of the data.
3.3.6 When were studies conducted?
Information on when guideline studies were executed gives insight into:
1. the extent to which REACH makes studies available, that already exisited, but
were not transparently available in one database;
2. how newly introduced alternative methods are taken up; and
3. how many new studies had to be done by registrants to ensure the safe use of
the chemicals on the market, where alternatives to the guideline testing were not
a viable option according to the registrants.
Figure 13 shows the distributions of the study period of the experimental studies (i.e.
when the study was carried out) as reported in the REACH registration database. For
each endpoint, the distribution is visualised as a boxplot, with the box showing the
quartiles (Q
1
=25 % and Q
3
=75 %) of the distribution.
The whiskers are drawn at 1.5 times the interquartile range (IQR=Q
3
-Q
1
) outside the low
and high quartiles. Points outside the whiskers are identified as outliers and drawn as
open circles. The vertical line within the boxplots corresponds to the median of the study
period distribution of the corresponding endpoint.
The annotations to the right of each boxplot show the number of unique “new” (2009
and later) and “old” (before 2009) studies. As in the previous version of this report,
2009 is taken as a significant point in time, as it defined the studies that generally
should be conducted and motivated by the REACH requirements – new studies that had
to be done by registrants to ensure the safe use and no viable alternative was available.
Other drivers such as other (global) legislative requirements cannot be excluded and will
also have contributed to the number of new studies executed.
The green vertical lines represent the different deadlines to give a more precise
orientation on how the REACH requirements may have influenced the behaviour of
registrants. Technical details on how the algorithms deduce the study period and on how
they establish the uniqueness of experimental studies can be found in Annex 1.

The use of alternatives to testing on animals
for the REACH Regulation
37
Figure 13: Distribution in the period of study per endpoint. “New” means studies
dated 2009 or later; “old” before 2009. The vertical green lines correspond to the
three REACH registration deadlines. See the text for a detailed explanation.
Figure 13 shows that REACH has brought transparency and availability to an enormous
collection of existing studies. The figure also illustrates that REACH seems to have
stimulated additional testing, revealing the existence of information gaps needed to
ensure safe use, where no other options were available.
In addition, the figure shows the evolvement towards integrated testing, and avoiding of
animal testing in general. An illustration of integrated testing is the use of the test

38
The use of alternatives to testing on animals
for the REACH Regulation
guideline combining 28-day repeated dose and reproductive toxicity screening (shown as
“combined 28d RDT with repro/dev” in Figure 13).
In 2016, 818 new studies (i.e. performed in or after 2009) were reported, along with
385 old ones. In 2019, 1 640 new ones were reported (an increase of 822), along with
432 old ones. This suggests that REACH, with most provisions starting to apply in 2008
and with the last registration deadline in 2018, has been the driver for most of the
studies combining 28-day repeated dose and reproductive toxicity screening.
Furthermore, REACH has also driven the use of in vitro skin corrosion/irritation, serious
eye damage/eye irritation and sensitisation tests. A striking example is the change for in
vitro serious eye damage/eye irritation studies, which quadrupled (from 677 to 2 635).
Similarly, in vitro skin corrosion/irritation tests tripled (from 1 291 to 3 642).
The most important increase is for in vitro skin sensitisation tests. In 2016, there were
only 67 tests, but in 2019, 1 322 tests were reported.
Figure 14 shows the occurrence of in vivo and in vitro studies for skin
corrosion/irritation, serious eye damage/eye irritation and sensitisation tests in time. It
illustrates that registrants choose more often the in-vitro route after the implementation
of the alternative in vitro method in the regulation as the first option; 2016 for in vitro
studies for skin corrosion/irritation as well as serious eye damage/eye irritation and 2017
for in vitro skin sensitisation.
Figure 14: Occurrence of in vivo and in vitro studies for skin corrosion/irritation,
serious eye damage/eye irritation and skin sensitisation tests over the years
1990- 2019

The use of alternatives to testing on animals
for the REACH Regulation
39
3.3.7 In-depth analysis: Skin corrosion/irritation and serious eye
damage/eye irritation
Since the last report, an increased amount of substances have been registered under
REACH, therefore the assessment of in vitro methods performed between 2016 and July
2019 will only focus on the number of in vitro studies performed without going into the
approaches used at substance level. In addition, no manual verification of the
compliance of the reported studies was performed due to the large amount of data
submitted.
For skin corrosion/irritation, most of the in vitro studies performed have been conducted
according to OECD TG 439 (In Vitro Skin Irritation: Reconstructed Human Epidermis Test
Method) or 431 (In vitro skin corrosion: reconstructed human epidermis (RHE) test
method). During this period, approximately 2 050 in vitro studies were performed.
Most of the studies were performed for substances registered between 1 and 100 tonnes
per year (ca. 1 950 studies). The majority (more than 65 %) of the studies were
performed according to OECD TG 439 for skin irritation, followed by OECD TG 431 for
skin corrosion. Fewer than 100 studies were performed according to OECD TGs 430 (In
Vitro Skin Corrosion: Transcutaneous Electrical Resistance Test) or 435 (In Vitro
Membrane Barrier Test Method).
For eye serious eye damage/eye irritation, most of the in vitro studies have been
conducted according to OECD TGs 437, 438 or 492. During this period, approximately
1 700 in vitro studies were performed. Again, the majority of the studies (ca. 1 150
studies) were performed for substances registered at tonnages between 1 and 100
tonnes per year. The Bovine Corneal Opacity and Permeability Test Method (OECD TG
437) was the most frequently used assay (ca. 900 studies performed), followed by OECD
TG 492 i.e. Reconstructed human Cornea-like Epithelium test method (ca. 600 studies
performed), and followed by OECD TG 438 i.e. the Isolated Chicken Eye test method
(ca. 230 studies performed). This demonstrates clearly that registrants are following the
amended information requirements and are only performing in vivo studies in
exceptional circumstances.
3.3.8 In-depth analysis: Skin sensitisation
Since the data mining performed for the previous report, the REACH information
requirements for skin sensitisation were amended and entered into force in May 2017.
The new legal requirements specify that if new data needs to be generated, the testing
would start with in vitro methods (covering three key events as described in the adverse
outcome pathway, OECD 2012). Only if the in vitro methods are not suitable for the
substance, or the results are not adequate for classification and, where required, for risk
assessment, can an in vivo study be performed.
Due to the 2018 registration deadline for the lower tonnage substances, an increased
number of in vitro studies for the skin sensitisation endpoint were generated for the
tonnage band of 1 to 100 tonnes per year. The whole of ECHA’s registration database
covering all tonnage bands currently contains approximately 1 500
34
studies for in vitro
methods among the different key events (55 studies were identified in the previous
report).
Most of the studies are covering inflammatory response in the keratinocyte key event
(40 %), followed by the molecular interaction with skin proteins key event (38 %) and
the activation of dendritic cells key event (22 %). For ca. 680 substances from all
34
This number i.e. ca. 1 500 studies differs from the number provided in Section 3.3.6 ( 1322 studies) as: i) in
vitro studies reported regardless of reliability were included, ii) non-standard in vitro studies were included, and
iii) multiple in vitro studies reported under one endpoint study record were calculated separately.

40
The use of alternatives to testing on animals
for the REACH Regulation
tonnage bands, data were generated using in vitro methods. Of those substances,
registrants are using information derived only from in vitro methods for about 70 % of
the substances (ca. 490 substances) to fulfil the information requirement and for 30 %
of the substances (ca. 190 substances) the information was derived from both in vitro
and in vivo methods.
The majority of the studies have been performed for substances registered at tonnages
from 1 to 100 tonnes per year (ca. 640 substances). The registration database seems to
indicate that following the amendment of the REACH standard information requirements
for skin sensitisation, registrants who needed to generate new information have started
testing using in vitro methods, where possible. Depending on the substance or results
obtained from the in vitro studies, in vivo testing (the Local Lymph Node Assay being the
preferred test method) may still be needed.
With the amendment of information requirements for skin sensitisation in 2017, it also
became mandatory to consider skin sensitisation potency for skin sensitising substances.
To this date, there is no internationally agreed way on how to do this based on in vitro
methods only. Therefore, currently this has to be done based on a weight-of-evidence
approach. To date, registrants have used the following approaches to estimate the skin
sensitisation potency of their substance by:
i) using in vitro study results only (for example, reactivity results based on OECD TG
442C or induction of cell surface markers at very low concentrations);
ii) using in vivo results obtained from a similar substance (Local Lymph Node Assay EC3
values) together with the in vitro study results; or
iii) using the QSAR Toolbox to estimate the potency based on analogue search together
with in vitro study results.
Under the OECD test guideline programme there is work ongoing to develop a guideline
for defined approaches for skin sensitisation. The guideline aims to provide a fixed data
interpretation procedure, to be used with a defined set of non-animal data, for the
identification of the skin sensitisation hazard, including the prediction of its potency.
3.4 Conclusions from the data analysis
With the inclusion of the 2018 phase-in substances, registered at the lower tier, the
coverage of substances in ECHA’s REACH registration database is complete. The
extensive analysis of the registration database, the availability of experimental studies
and the use of alternative options to fulfil the information requirements provide answers
to the questions posed earlier in this report:
What are the most commonly used adaptations?
A similar picture as in earlier editions of this report emerges. For all endpoints where
animal testing is or was the standard requirement, in practice, other means to fulfil
these requirements are more frequently used. This is with the exceptions of acute
toxicity, where for just over 50 % of the substances an in vivo testing approach was
used, mostly based on studies conducted before 2009.
For the more complex, higher tier endpoints, read-across is the preferred option to meet
the information requirements. In chapter 4 we will further discuss the quality of the
information submitted.
What are the most noticeable changes for higher-volume substances (100 tonnes per
year and above) since the previous (third) Article 117(3) report?
Again, a similar picture as in the earlier editions of this report can be observed. For
Annexes IX and X registrations, the overall situation did not change. For Annex X
substances there are hardly any changes detectable between 2016 and 2019. The

The use of alternatives to testing on animals
for the REACH Regulation
41
highest change, with an increase of 3 % for weight of evidence and a decrease of around
3 % for the use of data waivers, is observed in bioaccumulation. Developmental toxicity
– teratogenicity has seen a small increase in experimental studies (1.2 %), and an
increase in read-across (2.8 %), somewhat balanced with fewer testing proposals (-1.9
%) and less data waiving (-1.2 %).
For Annex IX, the overall picture is also very similar between 2016 and 2019 but some
changes are more clearly visible, especially for higher tier human health endpoints and
long-term toxicity to aquatic invertebrates. Developmental toxicity – teratogenicity has
shifted from testing proposal (5 % lower in 2019) to an increase in experimental studies
(7.1 % higher) in the 2016-2019 period. For repeated dose toxicity, an increase in
experimental tests of 3.1 % can be observed in the period of the analysis. Besides the
initiative by registrants (through testing proposals) to conduct studies for these
endpoints, these are also the endpoints that typically are requested after compliance
checks.
There has also been a slight increase in the percentage of experimental long-term
toxicity to fish studies (+1.4 % and +0.3 % for Annex IX and Annex X, respectively),
although the overall percentage of substances with experimental data for this endpoint
remains low (5 % and 6.1 % for Annex IX and X in 2019, respectively).
Which options did registrants use to fulfil their information requirements for lower-
volume substances (< 100 tonne per year)?
The general distribution of the adaptation options for Annex VII and VIII follows the
overall pattern observed previously for higher tier endpoints: read-across is the most
popular, followed by data waiving, weight of evidence and QSARs.
The 2019 Annex VIII dossiers have a similar use of adaptations as the 2019 Annex IX
dossiers, with the exception of acute toxicity where the Annex VIII dossiers have fewer
experimental studies (-3.1 %), which is compensated by an increase in the other options
(weight of evidence, QSAR and data waivers).
When comparing 2019 Annex VII dossiers (Figure 11) with 2019 Annex IX dossiers
(Figure 8) for the endpoints that are required at both tonnage bands, a generic trend can
be observed of fewer experimental studies and less read-across, balanced with more
weight of evidence, QSAR and data waiving. It can be concluded that for the dossiers
with the lowest data requirements, registrants have used alternative approaches, even
more than in the other tonnage bands.
Also remarkable is that for 37 % of all substances some information on the
bioaccumulation endpoint was submitted, even though bioaccumulation only needs to be
followed up at Annex VIII if screening information indicates a potential PBT concern. As
context, this information is present for roughly 50 % of the Annex IX and X substances.
Bioaccumulation does not need to be assessed for substances that have a low potential
for bioaccumulation (for example, based on a low octanol/water partition coefficient
(logK
ow
)) or a low potential to cross the biological membranes (in the case of high
molecular weight substances).
Finally, it was observed that for low tonnage substances the earlier registrations (2016)
had more additional information provided beyond the standard minimum requirements,
than those submitted more recently (2019). For substances in the lower tonnage bands
that are classified as carcinogenic, mutagenic or toxic to reproduction (CMR), categories
1 or 2, an earlier deadline applied (2009). It can be understood that these older dossiers
contain more information at a higher level than presently required by the tonnage band,
and that this led to their classification in the past.

42
The use of alternatives to testing on animals
for the REACH Regulation
How did the situation evolve for the endpoints where alternative methods have been
introduced since 2016 as standard information requirements?
For new and existing registrations, in vitro studies for skin corrosion/irritation, serious
eye damage/eye irritation and skin sensitisation have been clearly taken up since 2016.
The amendment of the REACH annexes has certainly played an important role in driving
this change.
4 Robustness of the used adaptations
In its General Report on the operation of REACH
35
, the European Commission
acknowledges that the development and consideration of alternative methods have
greatly improved during the last 10 years of REACH operation. The report also
acknowledges that registrants are attempting to minimise animal testing, while
recognising that some challenges still exist and that there are still gaps for some
endpoints – in particular, the high-tier ones.
The public consultation conducted for this report showed that while stakeholder views
concerning the achievements of REACH differed depending on their objectives, they were
consistently very positive concerning the promotion of alternative methods to animal
testing.
36
4.1 Testing strategies and adaptations
As can be seen from the data analysis, adaptations are used more often than guideline
studies. Registrants have used all options available to them, often combining many lines
of evidence. The underlying requirement is to ensure safe use through information
suitable for classification and risk assessment.
Unfortunately, many dossiers do not meet this requirement to ensure safe use as the
adaptations are not applied in a scientifically robust manner.
4.2 Use of read-across
Read-across is considered one of the main possible adaptations for higher tier human
health endpoints such as repeated dose toxicity, developmental and reproductive
toxicity, presuming that a scientifically plausible hypothesis can be justified and used to
derive a quantitative result for targeted substances.
If grouping and read-across are applied correctly, experimental testing can be reduced,
as there is no need to test every substance in a group for all required endpoints
37
.
However, experience from evaluation indicates that such adaptations provided by
registrants often fail to comply with the legal requirements and are inadequate to ensure
the safe use of chemicals. The most common shortcomings include:
• poor documentation, insufficient substance identification, significant deficiencies
in the quality of the source studies, lack of or low quality of supporting data;
• lack of qualitative and quantitative data to support predictions based on
toxicokinetics; and
• shortcomings in the hypothesis and justification of the toxicological prediction.
The deficiencies related to the supporting evidence are particularly relevant for high-tier
human health and high-tier environmental endpoints. To increase the robustness and
regulatory acceptance of those adaptations for high-tier human health endpoints,
additional data is needed, particularly related to toxicological mechanisms and
35
Commission General Report on the operation of REACH and review of certain elements
Conclusions and Actions
36
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52018SC0058&from=EN
37
https://echa.europa.eu/documents/10162/22931011/non_animal_approcches_en.pdf

The use of alternatives to testing on animals
for the REACH Regulation
43
absorption, distribution, metabolism and excretion (ADME) properties. The presence of
“mid-tier” screening studies like the 28-day repeated dose toxicity and/or screening for
reprotoxic effects can also strengthen the soundness of categories.
New approach methodologies (for example, high throughput in vitro screening) have the
potential to further substantiate the hypotheses of read-across approaches. As these
approaches often use starting points which are directly relevant for humans (such as
human liver cells), more relevant data can be obtained. One of the great challenges is
currently that these methods lack the integrated complexity of a higher organism
(metabolism, toxicokinetics, covering all effects etc.).
In March 2017, ECHA published the Read-Across Assessment Framework (RAAF)
38
. It
was first developed for human health endpoints and has later been extended to cover
environmental fate and effects, as well as considerations on multi-constituent and UVCB
substances.
The concepts of the RAAF were also incorporated in the OECD QSAR Toolbox. Although
the RAAF is an assessment framework intended for ECHA or Member State assessors, it
is a useful tool for registrants to self-assess the read-across/category developed in the
dossier, and also for strengthening their case to avoid testing all required endpoints for
all substances that are part of the read-across/category.
Based on the dossiers manually opened (for example, in compliance check), the
impression is that registrants have not in a significant number of cases taken the RAAF
as a guideline to proactively improve the read-across/category approaches in their
dossiers. This is one of the reasons that has led to an increased effort on compliance by
ECHA and the Commission.
39
4.1 Weight of evidence and data waiving
For weight of evidence and data waiving, experience from evaluation also indicates that
such adaptations provided by registrants are often found to be incompliant. Weight of
evidence and data waiving are often not supported by any reliable data or justification.
For weight of evidence, registrants often do not include reliable sources of information.
Moreover, ECHA’s evaluation experience indicates that, in most cases, weight of
evidence is not documented sufficiently (for example, the relevance of each line of
evidence is not described). In addition, registrants often do not ensure that each
element of the standard requirement is sufficiently covered in the proposed weight of
evidence.
To support registrants, ECHA published in 2017 a new reporting template to illustrate the
main required elements, with a background document on its use in human health and
environmental hazard assessments, in line with ECHA guidance.
40
4.2 Use of QSARs
In 2016, before the 2018 deadline, ECHA released a Practical Guide on How to use and
report (Q)SARs
41
, extended by practical examples on how to assess the reliability of
QSAR predictions with the most popular QSAR programs. For this report, ECHA
performed a manual screening assessment on a limited number of substances on the
quality of the information generated by QSAR models used by registrants as standalone
evidence. This use means that the information requirement was entirely covered by
38
https://echa.europa.eu/support/registration/how-to-avoid-unnecessary-testing-on-animals/grouping-of-
substances-and-read-across
39
https://echa.europa.eu/-/improving-compliance-is-echa-s-key-priority
40
https://echa.europa.eu/support/guidance-on-reach-and-clp-implementation/formats
41
https://echa.europa.eu/documents/10162/13655/pg_report_qsars_en.pdf

44
The use of alternatives to testing on animals
for the REACH Regulation
QSAR predictions (QSAR as the unique endpoint study record marked as “key study”).
Only dossiers submitted between 2016 and 2018 have been considered for this analysis.
The endpoints selected were short-term toxicity to fish, short-term toxicity to daphnia
and bioaccumulation. These endpoints have the highest percentage of QSAR predictions
used as standalone evidence, and if used adequately, reliable predictions can be made
due to the relatively high number of experimental data available and the lower levels of
complexity of the effect.
During the screening, adequacy of the input structure, applicability domain and the
coverage of the specific chemical space were verified. Overall, 75 predictions fulfilling the
criteria listed above were screened.
Out of the 50 QSAR predictions used as standalone information for short-term aquatic
toxicity, about one-third (32 %) were found not to be adequate in light of the conditions
for using QSAR predictions to adapt standard information requirements as listed in
REACH Annex XI, Article 1.3.
The most common reasons for non-reliable predictions were that the target substance
did not correspond to the registered substance, there was an insufficient number of data
points in the training set of the model, and a lack of coverage of the structural fragments
in the training set. Nevertheless, the remaining (68 %) QSAR predictions were found to
be adequate by this screening assessment, and accompanied by the appropriate QSAR
reporting format (QMRF and QPRF
42
) as required by REACH Annex XI, Article 1.3,
showing that QSARs can be a useful alternative to testing for short-term aquatic toxicity,
when used adequately.
Regarding bioaccumulation, 18 of the 25 selected substances had a logK
ow
above 3,
indicating a general potential for bioaccumulation in fat tissues. In our analysis, we
observed that more than two-thirds of these substances had shortcomings regarding the
assessment of the applicability of the model, which is a requirement for the use of QSAR
predictions as an adaptation to testing according to Annex XI, Article 1.3. While for one-
third of the substances, the reporting of the applicability domain check was limited to
logK
ow
and molecular weight (parametric domain), the other ca. 40 % of substances had
no applicability domain check reported at all.
To conclude, the majority of predictions for bioaccumulation (over 70 %) were found to
have shortcomings related to the applicability domain of the model. In addition, the
reliability of the prediction is often not sufficiently scrutinised by registrants. In
particular, this applies to the structural domain covered by the model, and the
anticipated biotransformation rate of the substance in cases where it significantly
reduces the predicted bioaccumulation potential of a substance.
The overall conclusion is that for aquatic toxicity, the QSAR approach as applied by the
registrants worked well in the majority of cases (68 %) while for bioaccumulation, the
majority (70 %) had issues.
42
QMRF = QSAR Model Reporting Format and QPRF = QSAR Prediction Reporting Format

The use of alternatives to testing on animals
for the REACH Regulation
45
5 Promotion of non-animal test methods
One of the objectives of REACH is to promote the use of alternative methods. To address
these challenges, ECHA is active in four areas:
1. Developing and maintaining tools and guidance (many in collaboration with the
OECD). ECHA has general as well as detailed guidance on the use of alternatives,
including specific guidance and manuals for specific topics (such as RAAF). In
addition, ECHA is the main financial contributor to the development of the OECD
QSAR Toolbox
43
, which is a tool that supports data gap filling through re-using
existing data (including the REACH registration data), as well as different
predictive approaches. A simplified and user-friendly version was released in
2020 with multiple new features helping users to obtain reliable predictions,
including category consistency assessment, automated workflow for skin
sensitisation and aquatic toxicity, and an exportable data matrix. Another
example is the practical guide released in July 2016 on how to use (Q)SARs,
which provides examples to assess the validity of predictions based on OECD
principles and REACH requirements.
44
2. Development and maintenance of OECD test guidelines and related
activities
45
. ECHA is (as part of the EU delegation) an active participant in the
OECD’s Working Group of National Coordinators of the TGs programme (WNT)
46
which develops test guidelines, the OECD harmonised templates to capture, share
and reuse test data electronically (IUCLID) and other activities to explore the
possibility of new approach methods in a regulatory context.
3. Exploring if and how new approach methods can be integrated in chemicals
management (international collaboration and OECD) mostly through APCRA.
47
4. Keeping ECHA staff up to date on the latest developments in
(eco)toxicology relevant in a regulatory context, through training, conferences,
expert fora and exchange (internal and external).
5.1 Building the knowledgebase of chemicals
As all substances on the European market should be registered by now, the focus of the
work has moved on from registration to aspects of REACH that ensure data are
compliant and are adequate to demonstrate safe use. The ultimate objective is to further
build and improve the IUCLID chemicals knowledgebase
48
, which will support avoiding
(additional) animal testing, the development of alternative methods, green/sustainable
chemistry, substitution and form a basis for the circular economy.
5.1.1 Further work needed by registrants
The further application and development of alternatives and the further development of
this chemicals knowledgebase is reliant on the receipt of updated and compliant
registrations that guarantee safe use, while utilising alternative methods in a sound
manner. This calls to mind some of the recommendations to registrants
49
in ECHA’s
43
https://www.oecd.org/chemicalsafety/risk-assessment/oecd-qsar-toolbox.htm
44
https://echa.europa.eu/practical-guides
45
https://www.oecd.org/env/ehs/testing/
46
https://www.oecd.org/env/ehs/testing/national-coordinators-test-guidelines-programme.htm
47
Accelerating the Pace of Chemical Risk Assessment
48
https://newsletter.echa.europa.eu/home/-/newsletter/entry/working-towards-one-global-iuclid How this data-
platform relates/is part of the “Feasibility study on a common open platform on chemical safety data” currently
executed by DG Environment is to be seen. https://etendering.ted.europa.eu/cft/cft-
document.html?docId=61946
49
https://echa.europa.eu/recommendations-to-registrants

46
The use of alternatives to testing on animals
for the REACH Regulation
annual reporting under Article 54 REACH,
50
particularly those on adaptation
possibilities.
51
Here it should be emphasised that in addition to in vitro methods, read-
across and QSAR are available as options to consider for avoiding testing on animals. In
the past decade, ECHA has clearly defined the conditions needed to construct read-
across/category and QSAR arguments that can withstand regulatory scrutiny and reliably
replace animal testing. These are crystallised in ECHA’s Practical Guide on How to use
and (Q)SARs and in the Read-Across Assessment Framework. If registrants choose to
use read-across and QSAR, they should take advantage of these to augment the
robustness of their adaptations.
5.1.2 Maximising the availability and use of data
Given the amount and complexity of the information collected, ECHA has developed
additional tools to help companies, authorities and researchers make the best use of it.
Since 2017, data from the dossiers has been made available for IUCLID users as a
downloadable file. This file contains all non-confidential substance data that has been
submitted to ECHA under REACH and reports study results for physical-chemical
properties, environmental fate and pathways, and (eco)toxicological information. The file
was last updated in April 2020
52
. This dataset has been integrated in the latest version of
the OECD QSAR Toolbox.
Besides the European efforts, many jurisdictions in the world have started to collect their
data using the same IUCLID software. Health Canada uses the IUCLID software as their
main scientific database for existing chemicals assessments. Switzerland and the US
have also started to accept data in IUCLID format for some parts of their chemical
legislation. Other countries, such as Australia and New Zealand, started using IUCLID
now that the tool is configured
53
to fit their regulatory contexts. This strengthens
interoperability among stakeholders and makes sharing and comparing practical.
The more widely the IUCLID format is accepted globally, the better this is for industry,
authorities and animal welfare organisations. It contributes to avoiding duplicate testing,
supporting international harmonisation of chemical data and reducing trade barriers.
Using a universal format based on IUCLID will in particular support the mutual
acceptance of test data since all authorities would ultimately have the same basis for
their assessments. In the long run, this would improve the efficiency of the work and
also increase the reliability of data which contributes significantly to the avoidance of
animal testing.
To really maximise the use of existing data, ECHA’s future vision is to support setting up
one international IUCLID platform: the Chemicals Knowledgebase. This would mean that
all parties involved, whether authorities or industry, would be able to contribute by
generating and entering data in the system.
Mutual acceptance of chemical safety data
54
is already a reality and having one
harmonised IT format for it might not be that far away. If and when this happens,
IUCLID has the potential to become the platform where chemical safety data can be
uploaded and viewed, but also managed, exchanged and improved by all parties. The
future platform would integrate the functionalities and options from other tools and
databases. For instance, in the coming years, the OECD QSAR Toolbox will be further
developed so that some of its key functionalities, like chemical similarity searches and
50
https://echa.europa.eu/overall-progress-in-evaluation
51
https://echa.europa.eu/adaptations-recommendations
52
https://iuclid6.echa.europa.eu/view-article/-/journal_content/title/reach-study-results-have-been-updated
53
http://www.oecd.org/chemicalsafety/risk-assessment/customisation-opportunities-of-iuclid-for-the-
management-of-chemical-data.pdf
54
https://www.oecd.org/env/ehs/mutualacceptanceofdatamad.htm

The use of alternatives to testing on animals
for the REACH Regulation
47
predicting hazard properties, could be made available through the Chemicals
Knowledgebase.
Currently, this is still a vision with a number of concrete building blocks operational.
ECHA will continue working towards this goal together with interested parties, such as
the OECD, which has recently agreed to further develop its OECD Global Chemicals
Knowledgebase in which the IUCLID Chemicals Knowledgebase will play a central role. In
the short term, the aim remains to make the data and knowledge gathered in the
framework of European chemicals legislation easier to access and use.
5.1.3 Using the REACH data for alternatives development: an example
With acute toxicity being one of the endpoints with the highest proportion of animal tests
in mind, ECHA collaborates with the US Interagency Coordinating Committee on the
Validation of Alternative Methods (ICCVAM)
55
. One of their priority projects is to develop
alternative methods for the “six-pack” tests; a set of basic tests – of which acute oral,
dermal and inhalation systemic toxicity tests are a part. While developing their models,
ECHA offered REACH registration data to extend their training and test sets. ECHA
supported ICCVAM by:
• investigating what acute oral toxicity data could be used for this purpose;
• extracting and filtering the data from ECHA’s database to provide data of
adequate quality for model development and validation; and
• giving advice to the model developers on the best possible use and interpretation
of the data.
When the models are finalised, they will be publicly available for all companies and
researchers to use free of charge. Discussions are ongoing on whether these models
could also be included in the QSAR Toolbox. Data exchange for the remaining endpoints
is also ongoing.
5.2 ECHA activities to promote the development of suitable
alternatives
REACH has as an objective to minimise the unnecessary use of animals in regulatory
hazard assessment. In addition, ECHA is facing many challenges, such as a large number
of incompliant dossiers, especially for higher tier endpoints, the need to improve
methodologies for risk assessment for ‘difficult’ scenarios (for example, substances with
complex compositions, mixture effects), the increasing expectations on high quality
information on chemicals that can be used to support policy objectives that move
towards using sustainable chemicals and the complexity of interpreting hazard data and
its translation to effective risk assessment and risk management measures.
ECHA is exploring ways to exploit new approach methodologies (NAMs) with the
ambition to test and demonstrate their applicability in a regulatory context. This
approach is envisaged to also enhance the pace of chemicals management, to have
better informed decisions and reduce or replace the need for studies on (vertebrate)
animals, for the protection of human health and the environment. The benefits of NAMs
should be observed in terms of:
• Throughput;
• Robustness;
• Bringing mechanistic knowledge;
55
https://ntp.niehs.nih.gov/whatwestudy/niceatm/iccvam/index.html

48
The use of alternatives to testing on animals
for the REACH Regulation
• Providing appropriate protection levels for human health and environment.
Recent ECHA reports that relate to this topic include the Integrated Regulatory Strategy
report,
56
the Applicability of non-animal approaches (ANAA) report (2017)
57
, the
proceedings of a scientific workshop on New Approach Methodologies in Regulatory
Science
58
, and the reporting under Article 54. With these in mind, ECHA’s regulatory and
scientific activities continue to promote non-animal test methods and testing strategies.
However, the conclusions from the ANAA
59
report remain valid. For complex endpoints,
such as repeated dose toxicity or reproductive toxicity, non-animal approaches are not
yet foreseeable. New approaches, such as in vitro microsystems and high-
throughput/high-content methods, are under development. They aim to provide better
insight into the mechanisms of toxicity. Still, they require further standardisation and
validation before they can be accepted for regulatory use. A continuous dialogue
between researchers and regulatory authorities is necessary to ensure that innovations
in non-animal approaches to chemical safety assessment can be considered for
regulatory use without undue delay.
5.3 Prospects for scientific development
As early as 2013, ECHA has stated in its Multi-Annual Work Programme 2014–2018:
“Significant and rapid development is being made, especially in (eco)toxicology, with an
emphasis on better understanding the biological mechanisms leading to an adverse
effect, rather than just observing the effect. Systems biology, bioinformatics, increased
understanding of modes of action and adverse outcome pathways will also affect the way
chemicals are tested, or how their properties can be predicted, thus enabling reduction
in traditional animal testing”.
Among the priorities outlined in its strategy, ECHA emphasised regulatory science
activities related to non-standard methods and new approaches methodologies to hazard
assessment, in particular rational integration of different lines of evidence (ITSs, IATAs,
AOPs;
60
with links to the QSAR Toolbox, omics and high-throughput screening
methodologies).
So far, most of the alternatives were developed by researchers with little attention to
their potential regulatory application. They were based mostly on in vitro systems. In
terms of opportunities, capturing mechanistic (for example, toxicokinetics, biomarkers)
data in parallel to adversities will allow to better understand the mode of action and
better predict potential adversities across species without actually relying on animal
data.
We see two main branches for development in the future:
For the short/medium term: a continuation of strengthening the ECHA knowledgebase.
An example is to use existing animal test systems (REACH standard information
requirement) and complement them when possible with multiple, relevant NAMs data.
The ambition is to bridge classical toxicological findings related to apical endpoints with
mechanistic knowledge. Moreover, this NAM-based information can work as bridging
studies, strengthening read-across and category arguments while reducing animal
testing.
For the long term: to predict systemic effects, new approaches need to cover a wide
range of tissues, organs, and chemical interactions within the organism (i.e.
56
https://echa.europa.eu/substances-of-potential-concern
57
https://echa.europa.eu/-/more-progress-needed-to-replace-animal-tests-under-eu-chemicals-laws
58
https://echa.europa.eu/documents/10162/22816069/scientific_ws_proceedings_en.pdf
59
https://echa.europa.eu/documents/10162/22931011/non_animal_approcches_en.pdf
60
Integrated testing strategy, integrated approach for testing and assessment, adverse outcome pathway.

The use of alternatives to testing on animals
for the REACH Regulation
49
toxicodynamics). Such a wide toxicological space cannot be covered by a single in vitro
assay. An intelligent combination of high throughput, high content assays and
computational tools could potentially fulfil these requirements. In addition to elements of
toxicodynamics, these methods need to cover toxicokinetics or ADME, namely:
Adsorption (estimates of systemic concentration), Distribution, Metabolism
(biotransformation) and Excretion.
It is still challenging nowadays to provide this wide coverage in both the toxicodynamic
and toxicokinetic aspects, as well as maintaining quality and throughput. The approach
presented in the United States Environmental Protection Agency’s research project
ToxCast, that deploys hundreds of high-throughput screening assays to generate
biological activity data, is trying to reach this ambitious goal, although it is premature to
estimate its impact on regulatory chemical safety.
61
In contrast to the well-developed understanding of the utility and limitations of NAM-
based biomarkers in the hazard assessment of pharmaceuticals, the practical utility of
these techniques for industrial chemicals is poorly understood. Another limiting factor is
that some industrial chemicals do not have suitable properties for in vitro testing (for
example, there are issues with solubility or volatility). To address this lack of
understanding, ECHA participates in an international consortium of regulatory agencies
“Accelerating the Pace of Chemical Risk Assessment” (APCRA), which is exploring ways
to use NAM-based information to inform the grouping of industrial chemicals in the
context of hazard assessment.
In the longer-term, APCRA is conducting a retrospective study to compare NAM-
delivered points of departure (mainly form high throughput assays) to those determined
by classical in vivo studies. The preliminary outcome of this study shows that in 92 % of
cases, NAMs can provide a conservative point of departure as protective or more
compared to classical in vivo data
62
. Based on this outcome, ECHA, together with
partners, continues to refine the methodology to show its utility in providing realistic
estimates for systemic toxicity, i.e. neither over-conservative nor under-protective. The
majority of substances in this part of the project are data-poor chemicals (i.e. for which
in vivo studies are not available).
With these actions, ECHA aims to cover both the short/medium-term and the long-term
prospects in NAM development, both making use of the huge amounts of toxicological
data, already available or being generated, and also by expanding and building on its
own knowledgebase.
61
https://www.epa.gov/chemical-research/toxcast-data-generation-chemical-workflow#phaseIII
62
Katie Paul Friedman, et al., Utility of In Vitro Bioactivity as a Lower Bound Estimate of In Vivo Adverse Effect
Levels and in Risk-Based Prioritization, Toxicological Sciences, Volume 173, Issue 1, January 2020, Pages 202–
225, https://doi.org/10.1093/toxsci/kfz201

50
The use of alternatives to testing on animals
for the REACH Regulation
6 Conclusions
For new and existing registrations, in vitro studies for skin corrosion/irritation, serious
eye damage/eye irritation and skin sensitisation have been clearly taken up since 2016.
The amendment of the REACH annexes has certainly played an important role in
accomplishing this significant change in the use of alternative methods.
In general, no major changes in the use of adaptations have been observed since
the last report in 2017
63
for the existing registrations. Furthermore, the following
observations can be made:
- Overall, the most commonly used adaptation is read-across, followed by
data waiving, weight of evidence and quantitative structure–activity relationship
(QSAR) models. Experimental studies were available – on average – in 27.1 % of
cases
(-0.5 % compared to 2016).
- When new studies are needed for repeated dose toxicity and toxicity to
reproduction screening, these are increasingly performed using the combined
repeated dose toxicity study with the reproduction/developmental toxicity
screening test (OECD 422). This significantly reduces the number of animals
and costs.
- There has been a moderate increase in the availability of pre-natal
developmental toxicity and (sub)chronic repeated dose studies. Decisions
related to compliance checks and testing proposals in the last three years are
likely to account for this.
In general, Annex VII and VIII dossiers received by the 2018 deadline follow the same
patterns in terms of use of adaptations. Furthermore, the following observations can be
made:
- The newly received 2019 Annex VIII dossiers follow a similar pattern as
dossiers in higher tonnage bands, with the exception of acute toxicity where
the Annex VIII dossiers have fewer experimental studies (-2.7 %), but weight of
evidence, QSAR and data waiving have increased. This seems to point to a lower
availabiltity of histrorical data, as well as an increased use of adaptations for this
group of substances.
- Remarkably, at REACH Annex VIII, the percentage of short-term toxicity to fish
studies used to fulfill the information requirement decreased since 2016, showing
an effective use of adaptations for this standard information requirement. In
addition, a minor increase for long-term aquatic experimental studies has been
observed.
- For newly received Annex VII dossiers, fewer experimental studies and
less read-across are observed, balanced with more weight of evidence,
QSAR and data waiving. For dossiers with the lowest data requirements, it can
be concluded that registrants have used alternative approaches, even more so
than in other tonnage bands. As these dossier have not been assed under
Compliance Check in significant number, it is for now unclear if the approaches
used are appropriate.
- Annex VII dossiers that were submitted earlier (before 2016) contain more
63
The data for the 2017 report was extracted in 2016, which is the date used throughout the report.

The use of alternatives to testing on animals
for the REACH Regulation
51
additional information on top of the standard minimum requirements than the
once submitted later (2019). Low tonnage substances also needed to be
registered before June 2010, if they were classified as carcinogenic, mutagenic or
toxic to reproduction, in categories 1 or 2 (CMR Cat 1 and 2). These substances,
can be expected to have more information than required according to the tonnage
band, since this information was likely forming the basis for their classification in
the past. Information that is needed to classify a substance as CMR Cat 1 and
2, is typically information that is only required starting from Annex IX. In
contrast, the substances that were registered later and are not classified as CMR
Cat 1 and 2, would not have this ‘extra’ information, as this is not required.
In terms of robustness of the applied alternatives, the picture of years before remains,
with frequent incompliances. This is despite the update and development of tools and
guidance, especially between the second (2013) and third deadline (2018).
As there are still many incompliances, many dossiers will need to undergo updates,
either voluntarily or after compliance check. Registrants still have opportunities to
strengthen their alternative approaches, based on ECHA Guidance and tools, as well the
feedback made available through other publications, such as the Article 54 reporting.
Despite the current issues with the robustness of the alternative approaches used in
registration dossiers, the REACH registration database, is a unique starting point for a
knowledgebase that can serve safe use of chemicals, sustainable chemistry
development, circular economy as well as the further development of alternative
approaches to animal testing. ECHA has developed a number of initiatives in this
direction and, stimulated by the emerging global acceptance of the IUCLID data standard
to capture and exchange study information, sees possibilities to develop such a
chemicals knowledgebase.
ECHA will continue to follow the developments at the OECD to seize opportunities to
bring alternative approaches into the regulatory context, as well as working together
with international partners in the APCRA
64
initiative to explore the use of more advanced
new approach methods.
64
https://www.epa.gov/chemical-research/accelerating-pace-chemical-risk-assessment-apcra

52
The use of alternatives to testing on animals
for the REACH Regulation
Annex 1
The purpose of this annex is to provide technical details on how the data analysis has
been carried out. Such technical details were kept intentionally out of the main body of
the report for brevity. The annex assumes that the reader is familiar with IUCLID and
the registration process, but reading it is not necessary for understanding the key
messages of the report. However, the annex may assist technical experts who wish to
know the conventions underpinning the data analysis and data visualisation, and it is
provided for completeness and transparency.
A1.1 Description of dossier and substance selection
The registration dossier list that was subjected to data analysis was constructed by
obtaining the submission of each registration that has the most recent submission date,
given that the submission date was on or before 31 July 2019. Both active and inactive
registrations were included in the data analysis. However, registrations that have been
revoked, annulled or invalidated were excluded.
65
Dossiers submitted by registrants that are members in the joint submission were
included in the analysis in case they provide fate and (eco) toxicity information not
present in the dossier submitted by the lead or individual registrants of the same
registered substance.
The analysis included NONS registrations that have been updated under REACH, given
that at least one dossier update has been received under REACH and that it has been
subject to full technical completeness check.
66
To analyse the evolution over time, we repeated the data analysis by applying a second
cut-off date, namely 31 July 2016 that roughly corresponds to the dataset used for the
purposes of the third Article 117(3) report. It is not possible to directly compare the
numbers included in this fourth Article 117(3) report with the numbers in the third
Article 117(3) report because of changes in the data analysis approach due to the
IUCLID update. The comparison of the results for the two cut-off dates nevertheless
provides an insight on the time evolution of how the information requirements have been
fulfilled.
Overall, we processed the data from 87 485 and 47 457 registrations for the 31 July
2019 and 31 July 2016 cut-off dates, respectively. The corresponding number of
substances can be found in Table 2.
Substances were allocated to REACH annexes according to the registration with the
highest information requirements at each point in time. As an illustration, a substance
has been considered as Annex IX if there is at least one REACH registration according to
Article 10 (so-called full registrations) with a tonnage of 100-1 000 tonnes per year and
no Article 10 registration with a higher tonnage.
The analysis covered only substances for which there is at least one registration that
provides all endpoint information as in Annex VII of REACH or higher. This means that
65
The registration status is not readily available as a function of time and, as such, registrations that were active
in the past and were annulled, invalid or revoked when the data analysis was carried out have been removed
even for result sets that refer to the past. This artefact has negligible effect on the obtained results.
66
NONs registrations pass through full technical completeness check if they have become the lead or they have
increased their tonnage band leading to increased information requirements compared to the original NONs
submission. It is likely that more NONs registrations have undergone full technical completeness check although
these criteria do not apply. Such NONs registrations were not included in the analysis. NONs registrations that
have not passed full technical completeness checks were excluded because of the incomplete migration of the
original NONS IUCLID dossiers to the latest IUCLID format that may skew the analysis.

The use of alternatives to testing on animals
for the REACH Regulation
53
substances for which registrants only provided the reduced information requirements
(physicochemical) of REACH Annex VII according to Article 12 1(b) were excluded
because such registrations typically do not contain tests on vertebrate animals or their
alternatives.
However, substances for which there has been at least one transported intermediate
registration for more than 1 000 tonnes per year have been included given that the
registrants are required to provide the full information requirements of REACH Annex
VII.
Once a substance was considered to be within the scope of the report then all
registrations were processed and analysed regardless of their own tonnage band.
A1.2 Processing of endpoint study records
This section of the annex summarises how individual endpoint study records were
extracted from the IUCLID database and processed. The next section describes how the
endpoint study record information has been aggregated at substance level to generate
the graphs.
The number of endpoint study records for the different IUCLID sections is shown in Table
4. The rows in this table correspond to the horizontal bars in the
barplots in Figures 4 - 11, i.e. for some endpoints, we counted together the endpoint
study records in more than one IUCLID section, as is for example the case for acute
toxicity where oral, inhalation and dermal studies are reported in separate IUCLID
sections but are counted here together.
Endpoint study records in category substances embedded in the registration dossiers
were excluded from the analysis, i.e. only the endpoint study records of the substance
that is the dossier subject were processed. Endpoint study records in IUCLID templates
embedded in the registered substance dataset were included in the analysis.
Table 4: Number of IUCLID endpoint study records in 2016 and 2019 dataset
Endpoint Number of
endpoint study
records in 2016
dataset
Number of
endpoint study
records in 2019
dataset
Increase (%)
bioaccumulation: aquatic
- sediment - terrestrial
21 014
29 498
40.37
short-term toxicity to
aquatic invertebrates
32 817
48 836
48.81
short-term toxicity to fish
28 539
39 552
38.59
long-term toxicity to
aquatic invertebrates
16 237
21 648
33.33
long-term toxicity to fish
11 395
14 547
27.66
toxicity to aquatic algae
and cyanobacteria
25 070
39 226
56.47
basic toxicokinetics -
dermal absorption
22 012
29 543
34.21

54
The use of alternatives to testing on animals
for the REACH Regulation
Endpoint Number of
endpoint study
records in 2016
dataset
Number of
endpoint study
records in 2019
dataset
Increase (%)
acute toxicity (all routes)
67 279
94 551
40.54
serious eye damage - eye
irritation
26 083
43 841
68.08
skin corrosion - irritation
36 120
55 534
53.75
skin sensitisation
23 094
44 009
90.56
genetic toxicity in vitro
50 733
75 191
48.21
genetic toxicity in vivo
13 661
17 941
31.33
repeated dose toxicity (all
routes)
48 092
66 329
37.92
developmental toxicity -
teratogenicity
18 077
26 460
46.37
toxicity to reproduction
15 070
23 852
58.27
carcinogenicity
11 008
12 621
14.65
Total
466 301
683 179
46.51
Table 4 shows that the number of endpoint study records increased by approximately
50 % between 2016 and 2019. The increase is primarily due to the last registration
deadline on 31 May 2018. The number of reliable, guideline experimental studies is
smaller than the number of endpoint study records shown because many endpoint study
records contain adaptations. Moreover, the same experimental study or adaptation may
have been reported in more than one endpoint study record in different dossiers for the
same or a different substance. For the purposes of this report, we have also attempted
to count the unique experimental studies as explained later on in this section.
With the introduction of IUCLID 6 there has been a significant change with regard to the
way registrants need to report read-across adaptations. While before registrants only
needed to provide one endpoint study record with the read-across information, with the
introduction of IUCLID 6, registrants are required to provide two endpoint study records,
one containing the experimental study with the source substance and one containing the
read-across adaptation for the registered substance that makes reference to the
endpoint study record with the source experimental study.
A side effect of this change is the fact that endpoint study records for which the type of
information has been indicated by the registrants to be an experimental study may refer
to an experiment carried out with a substance different to the one that has been
registered. This is one of the main reasons for which the data analysis approach
developed for the purposes of the third edition of the Article 117(3) report has been
modified.
A very large number of dossiers were submitted before the introduction of IUCLID 6 and
so the database contains all possible ways to report read-across and category

The use of alternatives to testing on animals
for the REACH Regulation
55
adaptations. For this reason, and in contrast to the methodology used for earlier editions
of the Article 117(3) report, judging whether an experimental study was carried out with
the registered substance or an analogue was not simple to establish using the
administrative information of the endpoint study records, so a more elaborate algorithm
was constructed. The main steps of this algorithm are shown in Table 5.
Table 5: Main elements of the algorithm to establish whether a test has been
carried out with the registered substance (test material algorithm)
Type of information Test material
matches
registered
substance
1
Endpoint study
record refers to
read-across
source
2
Algorithm
outcome
experimental study
no
it does not matter
read-across
no structured test
material information
yes
read-across
application
no structured test
material information
no
test with
registered
substance
yes
yes
read-across
application
yes
no
test with
registered
substance
migrated information:
read-across based on
grouping of substances
(category approach)
it does not matter
it does not matter
test with analogue
migrated information:
read-across from
supporting substance
(structural analogue or
surrogate)
it does not matter
it does not matter
test with analogue
read-across based on
grouping of substances
(category approach)
no
it does not matter
test with analogue
no structured test
material information
it does not matter
read-across
application
yes
it does not matter
read-across
application
read-across from
no
it does not matter
test with analogue

56
The use of alternatives to testing on animals
for the REACH Regulation
Type of information Test material
matches
registered
substance
1
Endpoint study
record refers to
read-across
source
2
Algorithm
outcome
supporting substance
(structural analogue or
surrogate)
no structured test
material information
it does not matter
read-across
application
yes
it does not matter
read-across
application
1
“yes” means that the test material contains at least one numerical identifier of the type EC
number or CAS number that matches the corresponding numerical identifier of the registered
substance, “no” means that the test material contains at least one identifier (e.g. a chemical
name) and neither the EC number nor CAS number (if contained) matches the corresponding
identifiers of the registered substance, “no structured test material information” means that the
test material does not contain any identifier (this can be an artefact of the automated migration
to IUCLID 6).
2
“yes” means that the type of information in the administrative part of the endpoint study
record is “experimental study” and the endpoint study record contains at least one cross
reference of the type “read-across source” for which the corresponding cross-referenced
document has been provided.
An endpoint study record was considered as an experimental study for the registered
substance if the test material matching the algorithm outcome was “test with registered
substance”. To count the percentage of substances with at least one experimental study
for the purposes of the barplots in Figures 4 - 11 and the circular plots in Figure 12 and
Figures in Annex 2, the endpoint study record should additionally have been identified as
reliable according to the registrant (Klimisch score 1 or 2, i.e. reliable without and with
restrictions, respectively) and, additionally, the study has been carried with one of the
guidelines mentioned later on in this section of the annex.
Most of the analysis and graphs presented in the report refer to the percentage of
substances that have an experimental study or for which a given adaptation has been
used. For these applications, it is not necessary to identify which endpoint study records
report the same original information. As an illustration, this often happens in cases of
read-across when both the source and the target substances of the read-across
adaptation have been registered. It is also frequent that the same experimental study is
used as the source in read-across adaptations for more than one registered substance.
For the purposes of the study period distributions shown in Figure 13, it was necessary
to count the unique experimental studies that have been identified as reliable according
to the registrant and executed according to one of the guidelines mentioned later in this
section of the annex. This was accomplished by creating study “signatures” in the form
of strings concatenating key information from the endpoint study record and, in
particular, from the study period, the guideline, the literature reference and the test
material.
Although more accurate signatures could have been created by using additional fields
from the endpoint study records, the benefit of the simple signatures is that they only
use fields that are present in all harmonised templates
(http://www.oecd.org/ehs/templates/
). This allows duplicate studies present in different
IUCLID sections to be detected, as can be the case, for example, for combined repeated
dose toxicity with reproduction/developmental toxicity screening studies.
It is important to emphasise though that any unique study identification algorithm based

The use of alternatives to testing on animals
for the REACH Regulation
57
on string equality like we used, may identify two endpoint study records that refer to the
same experimental study as distinct if one of the elements used to compile the signature
has been reported even slightly differently. This can, for instance, happen when in one of
the two literature references for the same study, the registrants provided the authors of
the study while in the other this information was missing although the bibliographic
reference may otherwise be identical. This suggests that Figure 13 overestimates the
number of unique studies. ECHA is currently investigating the possibility to use machine
learning for the purposes of un-duplicating studies, but this approach is still considered
experimental and not sufficiently developed to be used for the purposes of this report.
The study period distributions in Figure 13 also require a single characteristic date to be
computed that provides the time at which the experimental study has been conducted,
even though in reality the study took place over a period of time that can span several
months. A separate algorithm has been constructed to work out a single year that
roughly captures the time the study was conducted.
The algorithm uses all available sources of information and, in particular, the literature
reference year, the report date range and the study period provided by the registrant in
the administrative part of the endpoint record. The latter is a free text and dates were
extracted using natural language processing. From all dates, we only kept the year and
in cases of multiple extracted years, we retained only the latest that was used for
calculating the study period distributions visualised in the boxplots in Figure 13.
The last part of this section describes the way in which it was determined whether an
experimental study has been carried out with one of the generally acceptable guidelines.
As registrants may not always have used the IUCLID picklists, particularly for recently
developed in vitro methods that are important information for this report, we relied on
text pattern matching. The algorithm looked in all fields where guideline information may
have been provided. We ensured that the text patterns also correctly understood IUCLID
picklists if the registrants provided the guideline information in a structured manner,
which is the case for older studies for which guidelines have been available for several
years.
In some cases the same study has been tagged as matching more than one practically
equivalent guideline, for example, because the registrant provided both the EU and
OECD test guidelines. Such cases lead to the same study tag. In rare situations, the
registrants may have provided more than one guideline that lead two different study
tags, in which case the algorithm increased the study counts for both study tags.
Table 6 shows the assigned study tags (shown in Figures 1 - 3 and Figure 13), the
IUCLID sections where the study tags were applied to and the text patterns capturing
the generally acceptable guidelines. The text patterns are expressed in the form of
regular expressions and are only provided for transparency and completeness.
Table 6: Text patterns should for detecting guideline studies
Study tag IUCLID
section
Guideline
(sub)chronic RDT
7.5.1
(?i)(TG|OECD)[^0-9]*408([^\d]|$) or (?i)B[\.\s]*26([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+3100([^\d]|$) or (?i)(OPP)[^0-
9]*82[-\s]+1([^\d]|$) or (?i)(OTS)[^0-9]*798[\.\s]+2650([^\d]|$)
or (?i)(OTS)[^0-9]*795[\.\s]+2600([^\d]|$) or (?i)(TG|OECD)[^0-
9]*409([^\d]|$) or (?i)B[\.\s]*27([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3150([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+8700([^\d]|$) or (?i)(TG|OECD)[^0-9]*424([^\d]|$)
or (?i)B[\.\s]*43([^\d]|$)

58
The use of alternatives to testing on animals
for the REACH Regulation
Study tag IUCLID
section
Guideline
(sub)chronic RDT
7.5.1,
7.5.2,
7.5.3
(?i)(TG|OECD)[^0-9]*452([^\d]|$) or (?i)B[\.\s]*30([^\d]|$) or
(?i)(OPP)[^0-9]*83[-\s]+1([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+4100([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+3260([^\d]|$)
(sub)chronic RDT
7.5.2
(?i)(TG|OECD)[^0-9]*413([^\d]|$) or (?i)B[\.\s]*29([^\d]|$) or
(?i)(OPP)[^0-9]*82[-\s]+4([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3465([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+2450([^\d]|$)
(sub)chronic RDT
7.5.3
(?i)(TG|OECD)[^0-9]*411([^\d]|$) or (?i)B[\.\s]*28([^\d]|$) or
(?i)(OPP)[^0-9]*82[-\s]+3([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3250([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+2250([^\d]|$)
28d RDT
7.5.1
(?i)(TG|OECD)[^0-9]*407([^\d]|$) or (?i)B[\.\s]*7([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+3050([^\d]|$) or (?i)(TG|OECD)[^0-
9]*419([^\d]|$) or (?i)B[\.\s]*38([^\d]|$) or (?i)(OPP)[^0-9]*81[-
\s]+7([^\d]|$) or (?i)(OPP)[^0-9]*82[-\s]+5([^\d]|$) or
(?i)(OPP)[^0-9]*82[-\s]+6([^\d]|$)
28d RDT
7.5.2
(?i)(TG|OECD)[^0-9]*412([^\d]|$) or (?i)B[\.\s]*8([^\d]|$)
28d RDT
7.5.3
(?i)(TG|OECD)[^0-9]*410([^\d]|$) or (?i)B[\.\s]*9([^\d]|$) or
(?i)(OPP)[^0-9]*82[-\s]+2([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3200([^\d]|$)
acute toxicity
7.2.1
(?i)(TG|OECD)[^0-9]*401([^\d]|$) or (?i)(TG|OECD)[^0-
9]*420([^\d]|$) or (?i)(TG|OECD)[^0-9]*423([^\d]|$) or
(?i)(TG|OECD)[^0-9]*425([^\d]|$) or
(?i)B[\.\s]*1([^\d]|$)(?!bis)(?!tris) or (?i)B[\.\s]*1[-\.\s]*bis or
(?i)B[\.\s]*1[-\.\s]*tris or (?i)(OPP)[^0-9]*81[-\s]+1([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+1100([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+1175([^\d]|$)
acute toxicity
7.2.2
(?i)(TG|OECD)[^0-9]*403([^\d]|$) or (?i)(TG|OECD)[^0-
9]*433([^\d]|$) or (?i)(TG|OECD)[^0-9]*436([^\d]|$) or
(?i)B[\.\s]*2([^\d]|$) or (?i)B[\.\s]*52([^\d]|$) or (?i)(OPP)[^0-
9]*81[-\s]+3([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+1300([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+1150([^\d]|$)
acute toxicity
7.2.3
(?i)(TG|OECD)[^0-9]*402([^\d]|$) or (?i)(TG|OECD)[^0-
9]*434([^\d]|$) or (?i)B[\.\s]*3([^\d]|$) or (?i)(OPP)[^0-9]*81[-
\s]+2([^\d]|$) or (?i)(OPPTS)[^0-9]*870[\.\s]+1200([^\d]|$) or
(?i)(OTS)[^0-9]*798[\.\s]+1100([^\d]|$)
bioaccumulation
invertebrates
5.3.1
(?i)(OPPTS)[^0-9]*850[\.\s]+1710([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1830([^\d]|$) or (?i)(TG|OECD)[^0-9]*315([^\d]|$)
or (?i)(OPPTS)[^0-9]*835[\.\s]+4100([^\d]|$) or (?i)(OPPTS)[^0-
9]*835[\.\s]+4200([^\d]|$) or (?i)600[-/\s\\]+R[-/\s\\]+94[-
/\s\\]+024([^\d]|$) or (?i)(ASTM)[-/\s\\]+E[-/\s\\]*1688([^\d]|$)
or (?i)(OPP)[^0-9]*72[-\s]+6([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1850([^\d]|$)

The use of alternatives to testing on animals
for the REACH Regulation
59
Study tag IUCLID
section
Guideline
bioaccumulation
invertebrates
5.3.2
(?i)(TG|OECD)[^0-9]*317([^\d]|$) or (?i)C[\.\s]*30([^\d]|$) or
(?i)(ASTM)[-/\s\\]+E[-/\s\\]*1676([^\d]|$) or (?i)(ASTM)[-
/\s\\]+E[-/\s\\]*1688([^\d]|$)
bioaccumulation
vertebrates
5.3.1
(?i)(TG|OECD)[^0-9]*305([^\d]|$) or (?i)C[\.\s]*13([^\d]|$) or
(?i)(OPP)[^0-9]*165[-\s]+4([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1730([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1520([^\d]|$)
carcinogenicity
7.7
(?i)(TG|OECD)[^0-9]*451([^\d]|$) or (?i)B[\.\s]*32([^\d]|$) or
(?i)(OPP)[^0-9]*83[-\s]+2([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+3300([^\d]|$)
chronic/carcinogenici
ty
7.5.1,
7.5.2,
7.5.3, 7.7
(?i)(TG|OECD)[^0-9]*453([^\d]|$) or (?i)B[\.\s]*33([^\d]|$) or
(?i)(OPP)[^0-9]*83[-\s]+5([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+4300([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+3320([^\d]|$) or (?i)(OPP)[^0-9]*83[-\s]+5([^\d]|$)
combined 28d RDT
with repro/dev
screen
7.8.1,
7.8.2,
7.5.1,
7.5.2,
7.5.3
(?i)(TG|OECD)[^0-9]*422([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3650([^\d]|$)
developmental
toxicity
7.8.1,
7.8.2
(?i)(TG|OECD)[^0-9]*426([^\d]|$) or (?i)B[\.\s]*53([^\d]|$) or
(?i)(OPP)[^0-9]*83[-\s]+6([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+6300([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2500([^\d]|$)
developmental
toxicity
7.8.2
(?i)(TG|OECD)[^0-9]*414([^\d]|$) or (?i)B[\.\s]*31([^\d]|$) or
(?i)(OPP)[^0-9]*83[-\s]+3([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4900([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3600([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3700([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4350([^\d]|$)
eye
irritation/corrosion
(in vitro)
7.3.2
(?i)(TG|OECD)[^0-9]*437([^\d]|$) or (?i)B[\.\s]*47([^\d]|$) or
(?i)(TG|OECD)[^0-9]*438([^\d]|$) or (?i)B[\.\s]*48([^\d]|$) or
(?i)(TG|OECD)[^0-9]*491([^\d]|$) or (?i)(TG|OECD)[^0-
9]*492([^\d]|$) or (?i)(TG|OECD)[^0-9]*460([^\d]|$)
eye
irritation/corrosion
(in vivo)
7.3.2
(?i)B[\.\s]*5([^\d]|$) or (?i)(OPP)[^0-9]*81[-\s]+4([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+2400([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4500([^\d]|$) or (?i)(TG|OECD)[^0-9]*405([^\d]|$)
genetic toxicity (in
vitro)
7.6.1
(?i)(TG|OECD)[^0-9]*471([^\d]|$) or (?i)(TG|OECD)[^0-
9]*472([^\d]|$) or (?i)B[\.\s]*13([^\d]|$) or
(?i)B[\.\s]*14([^\d]|$) or (?i)(TG|OECD)[^0-9]*473([^\d]|$) or
(?i)B[\.\s]*10([^\d]|$) or (?i)(TG|OECD)[^0-9]*487([^\d]|$) or
(?i)(TG|OECD)[^0-9]*476([^\d]|$) or (?i)B[\.\s]*17([^\d]|$) or
(?i)(TG|OECD)[^0-9]*490([^\d]|$) or (?i)(TG|OECD)[^0-
9]*479([^\d]|$) or (?i)B[\.\s]*19([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+5900([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+5900([^\d]|$)

60
The use of alternatives to testing on animals
for the REACH Regulation
Study tag IUCLID
section
Guideline
genetic toxicity (in
vivo)
7.6.2
(?i)(TG|OECD)[^0-9]*474([^\d]|$) or (?i)B[\.\s]*12([^\d]|$) or
(?i)(TG|OECD)[^0-9]*475([^\d]|$) or (?i)B[\.\s]*11([^\d]|$) or
(?i)(TG|OECD)[^0-9]*488([^\d]|$) or (?i)B[\.\s]*58([^\d]|$) or
(?i)transgenic\s+(rodent|rat|mouse|mice) or (?i)(TG|OECD)[^0-
9]*489([^\d]|$) or (?i)Comet or (?i)(TG|OECD)[^0-
9]*485([^\d]|$) or (?i)B[\.\s]*25([^\d]|$) or (?i)(TG|OECD)[^0-
9]*483([^\d]|$) or (?i)B[\.\s]*23([^\d]|$) or (?i)(TG|OECD)[^0-
9]*478([^\d]|$) or (?i)B[\.\s]*22([^\d]|$) or (?i)dominant\s+lethal
or (?i)spermatogonial\s+chromosom(e|al)\s+aberration or
(?i)heritable\s+translocation
long-term toxicity to
aquatic
invertebrates
6.1.4
(?i)(TG|OECD)[^0-9]*211([^\d]|$) or (?i)(TG|OECD)[^0-
9]*242([^\d]|$) or (?i)(TG|OECD)[^0-9]*243([^\d]|$) or
(?i)C[\.\s]*20([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1300([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1350([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1330([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1950([^\d]|$)
long-term toxicity to
fish
6.1.2
(?i)(TG|OECD)[^0-9]*210([^\d]|$) or (?i)(TG|OECD)[^0-
9]*212([^\d]|$) or (?i)(TG|OECD)[^0-9]*215([^\d]|$) or
(?i)C[\.\s]*14([^\d]|$) or (?i)C[\.\s]*15([^\d]|$) or
(?i)C[\.\s]*47([^\d]|$) or (?i)(OPP)[^0-9]*72[-\s]+5([^\d]|$) or
(?i)(OPPTS)[^0-9]*850[\.\s]+1400([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1500([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1000([^\d]|$)
long-term toxicity to
fish
6.1.2,
6.1.4
(?i)(OPP)[^0-9]*72[-\s]+4([^\d]|$)
repro/dev toxicity
screening test
7.8.1,
7.8.2
(?i)(TG|OECD)[^0-9]*421([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3500([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4420([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+3550([^\d]|$)
short-term toxicity
to aqua. invert.
6.1.3
(?i)(TG|OECD)[^0-9]*202([^\d]|$) or (?i)(TG|OECD)[^0-
9]*235([^\d]|$) or (?i)C[\.\s]*2([^\d]|$) or (?i)(OPP)[^0-9]*72[-
\s]+2([^\d]|$) or (?i)(OPPTS)[^0-9]*850[\.\s]+1010([^\d]|$) or
(?i)(OPPTS)[^0-9]*850[\.\s]+1020([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1025([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1035([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1045([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1055([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+1200([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1300([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1800([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1930([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1970([^\d]|$) or (?i)(ISO)[^0-9]*6341([^\d]|$)

The use of alternatives to testing on animals
for the REACH Regulation
61
Study tag IUCLID
section
Guideline
short-term toxicity
to fish
6.1.1
(?i)(TG|OECD)[^0-9]*203([^\d]|$) or (?i)C[\.\s]*1([^\d]|$) or
(?i)(OPP)[^0-9]*72[-\s]+1([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1075([^\d]|$) or (?i)(OPPTS)[^0-
9]*850[\.\s]+1085([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1400([^\d]|$) or (?i)(OTS)[^0-
9]*797[\.\s]+1460([^\d]|$) or (?i)(ISO)[^0-9]*7346[-
\./\s\\]+1([^\d]|$) or (?i)C[\.\s]*49([^\d]|$) or (?i)(TG|OECD)[^0-
9]*236([^\d]|$)
short-term toxicity
to fish
6.1.1,
6.1.2
(?i)(TG|OECD)[^0-9]*204([^\d]|$)
short-term toxicity
to fish
6.1.1,
6.1.3
(?i)(OPP)[^0-9]*72[-\s]+3([^\d]|$)
skin
irritation/corrosion
(in vitro)
7.3.1
(?i)(TG|OECD)[^0-9]*430([^\d]|$) or (?i)(TG|OECD)[^0-
9]*431([^\d]|$) or (?i)reconstructed\s+human\s+epidermis or
(?i)(TG|OECD)[^0-9]*439([^\d]|$) or (?i)B[\.\s]*40([^\d]|$) or
(?i)transcutaneous\s+electrical\s+resistance or
(?i)human\s+skin\s+model or (?i)(TG|OECD)[^0-9]*435([^\d]|$)
or (?i)membrane\s+barrier\s+test or (?i)B[\.\s]*46([^\d]|$)
skin
irritation/corrosion
(in vivo)
7.3.1
(?i)(TG|OECD)[^0-9]*404([^\d]|$) or (?i)B[\.\s]*4([^\d]|$) or
(?i)(OPP)[^0-9]*81[-\s]+5([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4470([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+2500([^\d]|$)
skin sensitisation (in
vitro)
7.4.1
(?i)B[\.\s]*59([^\d]|$) or (?i)(TG|OECD)[^0-9]*442\s*C or DPRA
or (?i)direct peptide reactivity or (?i)in chemico skin sensiti.ation or
(?i)B[\.\s]*60([^\d]|$) or (?i)(TG|OECD)[^0-9]*442\s*D or
(?i)keratinosens or (?i)RE[-\s]*Nrf2\s+Luciferase or (?i)RE[-\s]*Nrf2
or (?i)LuSens or (?i)SENS[-\s]+IS or (?i)h[-\s]*CLAT or (?i)U[-
\s]+SENS or (?i)IL[-\s]*8[-\s-]*Luc
skin sensitisation (in
vivo)
7.4.1
(?i)(?<!non[-\s])LLNA or (?i)local\s+lymph\s*node or
(?i)B[\.\s]*42([^\d]|$) or (?i)(TG|OECD)[^0-9]*429 or GPMT or
(?i)guinea\s*pig\s+maximisation or (?i)B[\.\s]*6([^\d]|$) or
(?i)(TG|OECD)[^0-9]*406
toxicity to aqua.
algae and
cyanobact.
6.1.5
(?i)(TG|OECD)[^0-9]*201([^\d]|$) or (?i)C[\.\s]*3([^\d]|$) or
(?i)(OPP)[^0-9]*122[-\s]+2([^\d]|$) or (?i)(OPP)[^0-9]*123[-
\s]+3([^\d]|$) or (?i)(OPPTS)[^0-9]*850[\.\s]+5400([^\d]|$) or
(?i)(OTS)[^0-9]*797[\.\s]+1050([^\d]|$) or (?i)(ISO)[^0-
9]*8692([^\d]|$) or (?i)(ISO)[^0-9]*10253([^\d]|$)
toxicity to
reproduction
7.8.1
(?i)(TG|OECD)[^0-9]*415([^\d]|$) or (?i)B[\.\s]*34([^\d]|$) or
(?i)(TG|OECD)[^0-9]*443([^\d]|$) or (?i)B[\.\s]*56([^\d]|$) or
EOGRTS or (?i)extended[-\s]+one[-\s]+generation or
(?i)(TG|OECD)[^0-9]*416([^\d]|$) or (?i)B[\.\s]*35([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+3800([^\d]|$)
toxicity to
reproduction
7.8.1,
7.8.2
(?i)(OPP)[^0-9]*83[-\s]+4([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+4700([^\d]|$)

62
The use of alternatives to testing on animals
for the REACH Regulation
Study tag IUCLID
section
Guideline
toxicokinetics
7.1.1
(?i)(TG|OECD)[^0-9]*417([^\d]|$) or (?i)B[\.\s]*36([^\d]|$) or
(?i)(OPPTS)[^0-9]*870[\.\s]+8500([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2350([^\d]|$) or (?i)(OPP)[^0-9]*85[-\s]+1([^\d]|$)
or (?i)(OPPTS)[^0-9]*870[\.\s]+7485([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+8223([^\d]|$) or (?i)(OTS)[^0-
9]*798[\.\s]+7485([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+8360([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2230([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2280([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2300([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2310([^\d]|$) or
(?i)(transformation.dissolution.*metal.*aqueous\s+media|OECD
Series on Testing and Assessment.*\s+29([^\d]|$)) or
(?i)(OPPTS|OCSPP)[^0-9]*870[\.\s]+8245([^\d]|$)
toxicokinetics
7.1.1,
7.2.2
(?i)(OPPTS)[^0-9]*870[\.\s]+8320([^\d]|$) or (?i)(OPPTS)[^0-
9]*870[\.\s]+8380([^\d]|$) or (?i)(OTS)[^0-
9]*795[\.\s]+2320([^\d]|$)
A1.3 Aggregation of study information at substance level
This section of the annex builds on the previous one and summarises how the endpoint
study record information has been aggregated at substance level.
Contrary to earlier editions of the Article 117(3) report this edition does not include
multiple aggregation levels because they are not deemed necessary to convey the key
findings and are detrimental to readability. Instead, with the exception of Figure 13 that
displays the distribution of study periods without aggregating at substance level, all
other figures in the report have aggregated the endpoint study record information at
substance level, even when the endpoint study records have been retrieved from
different IUCLID dossiers for the same substance. Moreover, the figures can be
categorised into two main families:
• Figures 1 - 3 have grouped together the endpoint study records according to the
study tags assigned to them as described in the previous section. This means that
endpoint study records within the same IUCLID section may have been assigned
to different study tags because the IUCLID section encapsulates information that
refers to more than one information requirement as delineated in the REACH
annexes. As an example, this is the case for skin sensitisation where both in vitro
and in vivo studies are included in the same IUCLID section. Figures 1 - 3 only
examine the presence or absence of a study for each study tag for each
substance.
Figures 4 - 12, on the other hand, have grouped together the endpoint study records
according to the IUCLID section they belong. The technical reason for doing so is that
when the same IUCLID section encapsulates more than one information requirement, it
is technically challenging to assign all endpoint records in the same section to each
information requirement. For example, it is not always straightforward to algorithmically
assign a data waiver to a particular information requirement, especially for dossiers that
have not been recently updated. Such dossiers have automatically been migrated to the
latest IUCLID format and may not have passed the latest set of technical completeness
check rules that only started applying after their submission. For these reasons it has not

The use of alternatives to testing on animals
for the REACH Regulation
63
been technically possible to construct all figures so that the endpoint study records are
always grouped together according to the study tags, although this would have been
preferable for consistency.
A distinction is made between the endpoints on the vertical axis in red and in blue. The
endpoints in blue are required, depending on the relevant annex for the substance.
Some requirements are valid for all annexes (e.g. acute toxicity), some only occur at a
higher annex (repeated dose toxicity). To calculate the percentages, only the substances
in the corresponding annexes were used. Substances for which the endpoint was not a
requirement were left out. This was done, to provide a picture with the highest
resolution. Endpoints indicated in red are either optional or part of an integrated testing
approach and are not always required, regardless of the tonnage band. For this reason,
all categories have to be expressed versus all substances, regardless of the tonnage
bands. The category ‘no information’ is therefore relatively high.
The next part of this section explains how the information has been aggregated at
substance level for the purposes of the barplots in Figures 4 - 11. A cascade of rules was
applied after all endpoint study records in a given IUCLID section for the same substance
were pulled together regardless of whether they have been included in the same or
different IUCLID dossiers. All repeated dose toxicity IUCLID sections for the different
routes and duration were binned together. The same approach has been followed for
acute toxicity information, bioaccumulation and toxicokinetics. The aggregation rules can
be summarised as follows:
1. if there are no endpoint study records in the IUCLID section, the endpoint was
marked as “no information”; otherwise
2. if the only endpoint study records provided are one or more data waiver, the
endpoint was marked as “data waiver”; otherwise
3. if the only endpoint study records provided are one or more testing proposal the
endpoint was marked as “testing proposal”; otherwise
4. if at least one reliable (Klimisch score 1 or 2) experimental study with the
registered substance with one of the generally accepted guidelines under REACH
has been provided, the endpoint was marked as “experimental” regardless of the
presence of additional information; otherwise
5. if the only reliable (Klimisch score 1 or 2) information provided is one or more
read-across (but not reliable experimental study or QSAR prediction), the
endpoint was marked as “read-across/category”; otherwise
6. if the the only reliable (Klimisch score 1 or 2) information provided is one or more
QSAR prediction (but not reliable experimental study or read-across), the
endpoint was marked as “QSAR”; otherwise
7. if both reliable (Klimisch score 1 or 2) read-across and QSAR prediction
information has been provided (but no reliable experimental study), the endpoint
was marked as “weight of evidence”; otherwise
8. if the total number of unique endpoint study records that belong to one of the
following types:
o reliable experimental study with a generally not accepted guideline
o unreliable experimental study regardless of guideline
o unreliable read-across
o unreliable QSAR
o other information, not understood to be experimental study, read-across
or QSAR prediction
is two or more and there is there is no reliable experimental study, read-across
or QSAR prediction information, then endpoint was marked as “weight of
evidence”; otherwise
9. the endpoint was marked as “other”.

64
The use of alternatives to testing on animals
for the REACH Regulation
The above scheme is to some extent arbitrary and different hierarchy rules could have
been constructed. Despite this limitation, the use of hierarchical rules was deemed
essential to provide an overview that is otherwise impossible to convey if we enumerate
all possible combinations of endpoint study record types used to fulfil the information
requirements.
It is true that both the nature but also the sequence of hierarchical rules affect the
obtained results to some extent. However, the qualitative conclusions drawn in the
report would not differ significantly even with different rules. For this reason, and in the
interests of simplicity, this report does not contain results obtained with different sets of
hierarchical rules. Moreover, the effect of the adopted conventions is less significant
when the focus is on the way registrants have been changing the way they fulfil the
information requirements, given that both 2016 and 2019 datasets have been analysed
in a consistent way.
The next part of this section explains how the information has been aggregated at
substance level for the purposes of the circular graphs in Figure 12 and Figures in Annex
2. To some extent, these figures compensate for the shortcomings of the hierarchical
rules used for the barplots of Figures 4 - 11 by providing quantitative information on how
registrants combined different ways to fulfil the information requirements for the same
endpoint.
The colour coding of these circular graphs is consistent with the colour coding of the
barplots. To reduce the complexity of the graphs, we only show the percentage of
substances that:
1. have at least one endpoint study record that contains a reliable (Klimisch score 1
or 2) experimental study with the registered substance with one of the generally
accepted guidelines under REACH (dark blue);
2. have at least one endpoint study record that contains a reliable (Klimisch score 1
or 2) read-across (blue); or
3. have at least one endpoint study record that contains a reliable (Klimisch score 1
or 2) QSAR prediction (light blue).
All other endpoint study records have not been colour coded. However, this does not
mean that the corresponding IUCLID section is devoid of any hazard information. It is
possible that the combination of multiple pieces of evidence of lower reliability may be
sufficient to demonstrate the absence of hazard for some substances, or that the
registrant may have opted to conservatively assume that the substance is hazardous as
a worst case even though no definitive testing information is available.
In certain cases, this may be sufficient to ensure safe use if the classification and risk
assessment have also been conservatively applied.

The use of alternatives to testing on animals
for the REACH Regulation
65
Annex 2
A2.1 Detailed overviews of options used for each endpoint,
covering all tonnage bands
This annex contains additional graphs from Section 3.3.5 that have been omitted from
the main body of the report for brevity.
Figure 15. Detailed view for environmental toxicity and fate endpoints 2019
a) Bioaccumulation

66
The use of alternatives to testing on animals
for the REACH Regulation
b) Short-term toxicity to aquatic invertebrates [short-term toxicity to aquatic
invertebrates_2019]
c) Short-term toxicity to fish
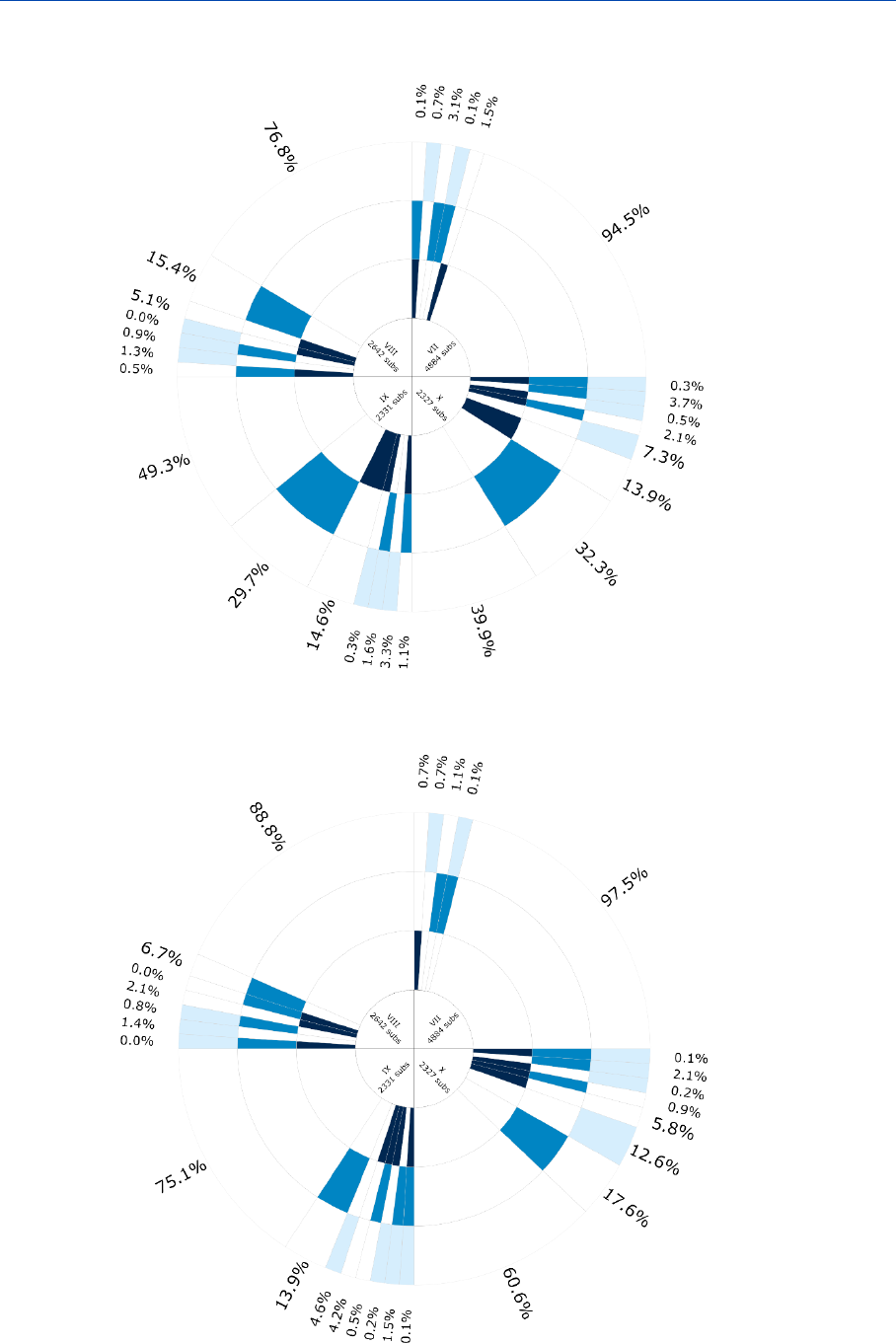
The use of alternatives to testing on animals
for the REACH Regulation
67
d) Long-term toxicity to aquatic invertebrates
e) Long-term toxicity to fish

68
The use of alternatives to testing on animals
for the REACH Regulation
f) Toxicity to aquatic algae and cyanobacteria
Figure 16. Detailed view for human health lower tier endpoints 2019
a) Acute toxicity (all routes)

The use of alternatives to testing on animals
for the REACH Regulation
69
b) Serious eye damage - eye irritation
c) Skin corrosion - irritation

70
The use of alternatives to testing on animals
for the REACH Regulation
d) Skin sensitisation
e) Genetic toxicity in vitro
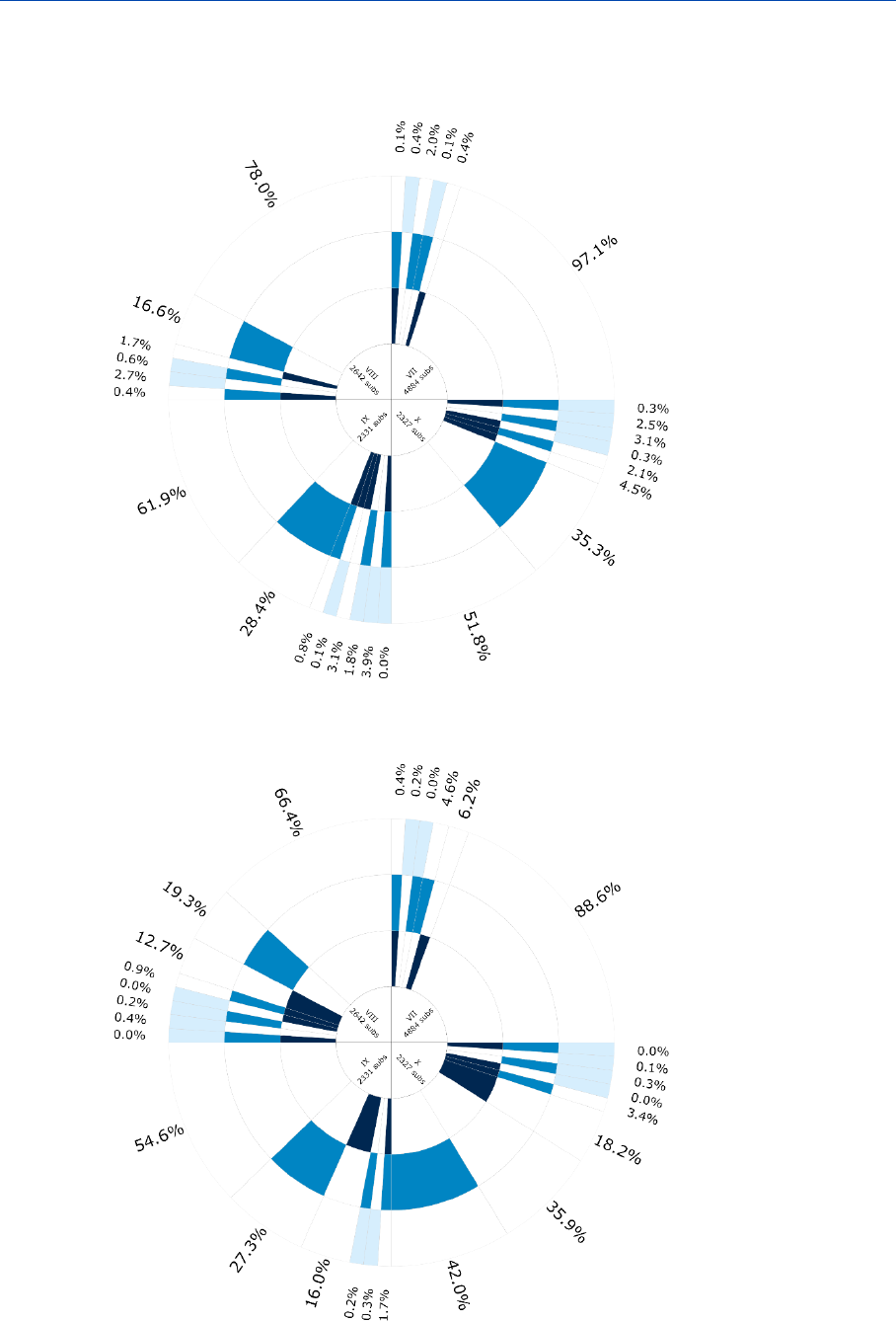
The use of alternatives to testing on animals
for the REACH Regulation
71
Figure 17. Detailed view for human health higher tier endpoints 2019
a) Basic toxicokinetics - dermal absorption
b) Genetic toxicity in vivo

72
The use of alternatives to testing on animals
for the REACH Regulation
c) Repeated dose toxicity (all routes)
d) Developmental toxicity – teratogenicity

The use of alternatives to testing on animals
for the REACH Regulation
73
e) Toxicity to reproduction
f) Carcinogenicity
74
The use of alternatives to testing on animals
for the REACH Regulation
Annex 3
A3.1 Detailed results of the options analysis
This annex contains the detailed results of the option analysis, used to make the figures
4 – 11 in chapter 3.3.2 – 3.3.5
Table 7: Frequency of the different options to fulfil the information
requirements in 2019 (aggregated at IUCLID section level), relates to Figure 4.
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 2.7 9.8 0.6 1.9 6.8 0.0 2.4 75.7
toxicity to reproduction 16.3 30.3 1.1 2.0 11.4 0.4 3.0 35.5
developmental
/teratogenicity
14.1 27.3 0.8 1.5 6.0 0.9 2.0 47.4
repeated dose toxicity
(all routes)
27.7 33.9 0.8 2.0 1.4 0.1 5.0 29.0
genetic toxicity in vivo 13.1 19.3 0.2 1.7 2.8 0.7 3.1 58.9
genetic toxicity in vitro 54.7 33.2 1.5 6.5 1.1 0.0 1.7 1.2
skin sensitisation 42.2 35.1 3.2 4.2 7.3 0.0 6.7 1.4
skin corrosion -
irritation
50.4 31.4 1.8 3.2 3.3 0.0 8.5 1.3
serious eye damage -
eye irritation
47.4 31.3 1.8 2.9 6.2 0.0 9.1 1.3
acute toxicity (all
routes)
52.5 32.1 1.4 6.4 2.1 0.0 4.3 1.2
basic toxicokinetics -
dermal absorption
2.8 16.6 2.0 6.2 5.2 0.0 14.6 52.7
toxicity to aquatic algae
and cyanobacteria
43.6 33.8 4.4 4.6 6.0 0.0 4.5 3.0

The use of alternatives to testing on animals
for the REACH Regulation
75
Study Read-
across
QSAR WoE Waiving TP Other No info
long-term toxicity to
fish
3.1 7.9 3.9 1.3 27.0 0.2 2.0 54.7
long-term toxicity to
aquatic invertebrates
8.1 16.4 2.6 1.8 16.8 0.4 2.0 51.9
short-term toxicity to
fish
31.8 27.9 3.1 4.8 2.6 0.0 5.6 24.3
short-term toxicity to
aquatic invertebrates
46.2 33.8 3.9 5.0 4.5 0.0 5.3 1.3
bioaccumulation 3.4 7.1 11.5 6.6 21.3 0.1 2.7 47.4
Table 8: Frequency of the different options to fulfil the information
requirements in 2016 (aggregated at IUCLID section level), relates to Figure 5
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 3.7 14.4 0.9 2.5 10.5 0.0 3.5 64.6
toxicity to reproduction 16.6 34.9 1.4 1.8 14.7 0.7 3.5 26.5
developmental
/teratogenicity
16.2 33.9 1.2 1.9 9.7 3.2 2.8 31.2
repeated dose toxicity
(all routes)
31.5 38.4 1.0 2.1 1.9 0.3 6.8 17.9
genetic toxicity in vivo 16.1 24.9 0.2 2.0 3.9 0.6 3.6 48.6
genetic toxicity in vitro 53.5 33.4 2.1 5.9 1.5 0.0 1.4 2.3
skin sensitisation 41.0 36.0 2.5 3.0 8.1 0.0 6.7 2.5
skin corrosion -
irritation
48.1 31.3 1.8 3.8 4.2 0.0 8.5 2.3

76
The use of alternatives to testing on animals
for the REACH Regulation
Study Read-
across
QSAR WoE Waiving TP Other No info
serious eye damage -
eye irritation
44.1 31.6 1.5 3.3 7.3 0.0 9.7 2.4
acute toxicity (all
routes)
53.4 31.2 1.7 5.8 1.9 0.0 3.7 2.3
basic toxicokinetics -
dermal absorption
3.4 21.7 2.0 8.4 7.3 0.0 16.5 40.7
toxicity to aquatic algae
and cyanobacteria
42.4 34.6 4.4 4.1 5.0 0.0 6.4 3.2
long-term toxicity to
fish
3.7 10.2 5.1 1.3 41.7 0.3 2.3 35.4
long-term toxicity to
aquatic invertebrates
10.0 20.8 3.0 2.3 26.6 0.7 3.1 33.5
short-term toxicity to
fish
36.4 31.0 3.2 5.5 2.6 0.0 7.2 14.1
short-term toxicity to
aquatic invertebrates
44.5 33.6 4.0 4.4 4.7 0.0 6.5 2.3
bioaccumulation 4.1 9.0 14.6 5.6 32.2 0.1 3.7 30.7
Table 9: Frequency of the different options to fulfil the information
requirements for Annex X substances in 2019 (aggregated at IUCLID section
level), relates to Figure 6
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 8.0 30.9 1.8 4.0 24.9 0.0 5.7 24.6
toxicity to reproduction 21.2 53.3 1.8 1.3 16.4 1.5 4.1 0.3

The use of alternatives to testing on animals
for the REACH Regulation
77
Study Read-
across
QSAR WoE Waiving TP Other No info
developmental
/teratogenicity 28.8 53.1 1.6 1.6 11.3 0.6 2.6 0.3
repeated dose toxicity
(all routes) 39.0 54.0 1.6 1.1 1.7 0.0 2.3 0.3
genetic toxicity in vivo 21.7 42.0 0.1 2.7 6.3 0.1 3.3 23.9
genetic toxicity in vitro 50.7 44.6 1.6 1.4 0.9 0.0 0.5 0.3
skin sensitisation 36.7 46.0 1.9 2.8 8.3 0.0 4.1 0.3
skin corrosion -
irritation 45.3 40.4 1.3 2.3 4.2 0.0 6.2 0.3
serious eye damage -
eye irritation 42.1 41.7 1.4 2.1 6.2 0.0 6.3 0.3
acute toxicity (all
routes) 53.6 39.6 1.4 1.2 1.0 0.0 2.8 0.3
basic toxicokinetics -
dermal absorption 7.3 35.3 2.5 14.3 11.3 0.0 10.9 18.4
toxicity to aquatic algae
and cyanobacteria 40.8 41.7 4.6 6.7 3.0 0.0 2.8 0.3
long-term toxicity to
fish 7.1 17.6 12.6 3.1 54.3 0.4 4.5 0.3
long-term toxicity to
aquatic invertebrates 16.8 32.3 7.3 5.0 33.4 0.7 4.2 0.3
short-term toxicity to
fish 41.7 39.4 4.4 8.6 2.9 0.0 2.6 0.3
short-term toxicity to
aquatic invertebrates 42.2 40.6 4.2 7.0 3.2 0.0 2.4 0.3
78
The use of alternatives to testing on animals
for the REACH Regulation
Study Read-
across
QSAR WoE Waiving TP Other No info
bioaccumulation 6.1 12.7 17.3 10.7 47.6 0.0 5.2 0.3
Table 10: Frequency of the different options to fulfil the information
requirements for Annex X substances in 2016 (aggregated at IUCLID section
level), relates to Figure 7
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 7.9 31.1 1.8 3.9 24.7 0.0 6.3 24.2
toxicity to reproduction 20.3 52.3 1.5 1.9 17.6 1.6 4.0 0.8
developmental
/teratogenicity 27.6 50.4 1.5 1.6 12.5 2.5 3.2 0.7
repeated dose toxicity
(all routes) 39.4 52.4 1.5 1.2 2.3 0.1 2.4 0.7
genetic toxicity in vivo 21.8 41.8 0.1 2.5 7.2 0.3 3.3 22.9
genetic toxicity in vitro 51.4 43.4 1.5 1.5 1.0 0.0 0.5 0.7
skin sensitisation 37.1 45.1 1.7 3.6 8.7 0.0 3.2 0.7
skin corrosion -
irritation 46.5 38.4 1.3 4.5 4.3 0.0 4.4 0.7
serious eye damage -
eye irritation 42.4 40.1 1.3 4.3 6.3 0.0 4.8 0.7
acute toxicity (all
routes) 54.0 38.6 1.3 1.3 1.0 0.0 3.0 0.7
basic toxicokinetics -
dermal absorption 6.8 34.2 1.5 14.7 11.9 0.0 12.4 18.4

The use of alternatives to testing on animals
for the REACH Regulation
79
Study Read-
across
QSAR WoE Waiving TP Other No info
toxicity to aquatic algae
and cyanobacteria 40.6 40.7 4.7 6.7 3.3 0.0 3.2 0.7
long-term toxicity to
fish 6.9 16.9 12.7 2.7 55.9 0.1 4.1 0.8
long-term toxicity to
aquatic invertebrates 15.7 31.8 6.9 4.7 35.2 0.6 4.4 0.7
short-term toxicity to
fish 41.8 38.6 4.4 8.7 3.0 0.0 2.8 0.7
short-term toxicity to
aquatic invertebrates 42.5 39.2 4.3 7.1 3.8 0.0 2.4 0.7
bioaccumulation 5.7 11.9 17.8 7.3 50.9 0.2 5.5 0.8
Table 11: Frequency of the different options to fulfil the information
requirements for Annex IX substances in 2019 (aggregated at IUCLID section
level), relates to Figure 8
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 2.8 11.9 0.6 2.4 7.4 0.0 3.2 71.8
toxicity to reproduction 24.1 46.3 0.9 1.4 21.8 0.3 4.7 0.4
developmental
/teratogenicity 26.3 49.9 0.7 1.9 14.4 3.3 2.8 0.6
repeated dose toxicity
(all routes) 40.2 51.7 0.5 1.2 2.0 0.4 3.6 0.4
genetic toxicity in vivo 17.7 27.3 0.3 2.1 4.8 0.4 2.3 45.1
genetic toxicity in vitro 59.8 36.3 0.8 1.3 0.9 0.0 0.4 0.4

80
The use of alternatives to testing on animals
for the REACH Regulation
Study Read-
across
QSAR WoE Waiving TP Other No info
skin sensitisation 45.2 40.4 1.5 2.4 6.5 0.0 3.6 0.4
skin corrosion -
irritation 54.5 34.5 0.8 1.5 3.0 0.0 5.2 0.4
serious eye damage -
eye irritation 50.5 33.6 0.9 1.8 6.3 0.0 6.6 0.4
acute toxicity (all
routes) 59.8 33.8 0.6 1.2 1.3 0.0 2.9 0.4
basic toxicokinetics -
dermal absorption 4.0 28.4 3.9 7.4 7.4 0.0 16.7 32.2
toxicity to aquatic algae
and cyanobacteria 46.4 41.8 2.1 3.2 3.4 0.0 2.6 0.6
long-term toxicity to
fish 5.0 13.9 4.6 1.8 71.6 0.6 2.0 0.5
long-term toxicity to
aquatic invertebrates 16.1 29.7 3.3 2.1 45.0 0.8 2.5 0.5
short-term toxicity to
fish 43.7 43.2 2.4 3.7 2.9 0.0 3.6 0.4
short-term toxicity to
aquatic invertebrates 47.9 39.5 1.8 2.7 4.4 0.0 3.3 0.4
bioaccumulation 5.0 12.6 19.6 9.7 48.2 0.2 4.1 0.5

The use of alternatives to testing on animals
for the REACH Regulation
81
Table 12: Frequency of the different options to fulfil the information
requirements for Annex IX substances in 2016 (aggregated at IUCLID section
level), relates to Figure 9
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 2.6 12.1 0.5 2.2 8.6 0.0 3.1 71.0
toxicity to reproduction 22.1 46.5 1.0 1.9 22.9 0.7 4.1 0.8
developmental
/teratogenicity 19.2 48.9 0.9 2.0 16.6 8.3 3.1 1.0
repeated dose toxicity
(all routes) 37.1 52.7 0.7 1.7 2.3 0.8 4.0 0.7
genetic toxicity in vivo 16.4 27.4 0.4 1.8 4.7 1.1 2.3 46.1
genetic toxicity in vitro 59.6 35.3 1.2 1.8 1.1 0.0 0.3 0.7
skin sensitisation 44.6 40.2 1.9 2.4 7.1 0.0 3.2 0.7
skin corrosion -
irritation 54.4 33.5 1.0 2.8 3.5 0.0 4.2 0.7
serious eye damage -
eye irritation 50.3 33.3 1.1 2.4 6.7 0.0 5.6 0.7
acute toxicity (all
routes) 60.2 32.3 0.8 1.3 1.4 0.0 3.2 0.7
basic toxicokinetics -
dermal absorption 3.3 29.3 3.4 7.7 8.7 0.0 15.7 31.9
toxicity to aquatic algae
and cyanobacteria 45.6 41.2 2.6 3.1 3.6 0.0 2.9 1.0
long-term toxicity to
fish 3.6 13.3 3.2 1.0 75.2 0.9 2.0 0.9
long-term toxicity to
aquatic invertebrates 13.0 28.8 2.2 1.6 48.5 1.9 3.0 0.9

82
The use of alternatives to testing on animals
for the REACH Regulation
Study Read-
across
QSAR WoE Waiving TP Other No info
short-term toxicity to
fish 43.5 42.7 2.4 3.8 3.4 0.0 3.5 0.7
short-term toxicity to
aquatic invertebrates 47.9 38.2 2.3 3.0 4.7 0.0 3.2 0.7
bioaccumulation 4.6 12.3 19.5 8.6 50.0 0.2 3.8 0.9
Table 13: Frequency of the different options to fulfil the information
requirements for Annex VIII substances in 2019 (aggregated at IUCLID section
level), relates to Figure 10
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 1.6 5.2 0.5 1.2 1.6 0.0 1.4 88.5
toxicity to reproduction 31.4 44.3 0.9 2.4 16.6 0.2 3.4 0.7
developmental
/teratogenicity 12.5 29.9 0.5 2.0 3.5 0.8 2.0 48.8
repeated dose toxicity
(all routes) 39.7 51.1 0.8 1.7 2.4 0.2 3.7 0.4
genetic toxicity in vivo 13.7 19.3 0.4 1.2 2.0 2.0 2.5 58.9
genetic toxicity in vitro 61.0 32.7 0.9 3.6 1.0 0.0 0.4 0.4
skin sensitisation 46.3 37.0 2.0 3.2 7.0 0.0 4.1 0.4
skin corrosion -
irritation 55.7 31.9 1.1 1.6 3.1 0.0 6.2 0.4
serious eye damage -
eye irritation 52.9 31.6 1.1 1.2 6.7 0.0 6.1 0.4

The use of alternatives to testing on animals
for the REACH Regulation
83
Study Read-
across
QSAR WoE Waiving TP Other No info
acute toxicity (all
routes) 56.7 34.2 0.9 1.8 1.8 0.0 4.2 0.4
basic toxicokinetics -
dermal absorption 2.1 16.6 2.7 5.2 5.9 0.0 25.1 42.4
toxicity to aquatic algae
and cyanobacteria 46.4 36.9 3.1 2.9 6.5 0.0 2.5 1.7
long-term toxicity to
fish 2.2 6.7 1.4 1.1 10.0 0.1 1.6 77.0
long-term toxicity to
aquatic invertebrates 5.6 15.4 1.3 1.0 5.8 0.3 1.1 69.5
short-term toxicity to
fish 44.2 39.8 3.9 3.5 4.4 0.0 3.7 0.4
short-term toxicity to
aquatic invertebrates 49.9 36.3 2.5 3.5 4.8 0.0 2.6 0.4
bioaccumulation 3.3 7.2 11.0 4.0 8.7 0.1 2.0 63.7
Table 14: Frequency of the different options to fulfil the information
requirements for Annex VII substances in 2019 (aggregated at IUCLID section
level), relates to Figure 11
Study Read-
across
QSAR WoE Waiving TP Other No info
carcinogenicity 0.6 1.3 0.2 1.1 0.7 0.0 1.1 95.0
toxicity to reproduction 2.2 4.0 1.0 2.4 1.3 0.0 1.4 87.7
developmental
/teratogenicity 2.0 2.9 0.5 1.0 0.9 0.0 1.3 91.4
repeated dose toxicity
(all routes) 9.8 6.5 0.6 3.0 0.6 0.0 7.8 71.7

84
The use of alternatives to testing on animals
for the REACH Regulation
Study Read-
across
QSAR WoE Waiving TP Other No info
genetic toxicity in vivo 6.6 4.6 0.2 1.4 0.7 0.4 3.7 82.3
genetic toxicity in vitro 50.8 26.7 2.2 12.9 1.2 0.0 3.7 2.6
skin sensitisation 41.3 26.2 5.2 6.3 7.3 0.0 10.7 2.9
skin corrosion -
irritation 48.1 25.5 2.9 5.2 3.3 0.0 12.5 2.6
serious eye damage -
eye irritation 45.5 25.1 2.8 4.8 5.9 0.0 13.2 2.7
acute toxicity (all
routes) 46.3 26.5 2.1 13.8 3.1 0.0 5.7 2.6
basic toxicokinetics -
dermal absorption 0.4 2.0 0.4 2.3 0.8 0.0 9.6 84.5
toxicity to aquatic algae
and cyanobacteria 42.2 24.5 6.1 5.3 8.4 0.0 7.3 6.2
long-term toxicity to
fish 0.7 1.1 0.7 0.3 2.0 0.0 1.0 94.3
long-term toxicity to
aquatic invertebrates 1.6 3.1 0.7 0.6 1.3 0.0 1.3 91.4
short-term toxicity to
fish 14.7 8.6 2.4 4.1 1.2 0.0 8.9 60.0
short-term toxicity to
aquatic invertebrates 45.2 26.5 5.7 6.0 5.0 0.0 9.0 2.6
bioaccumulation 1.4 1.7 5.2 4.5 2.6 0.0 1.1 83.5
EUROPEAN CHEMICALS AGENCY
P.O. BOX 400, FI-00121 HELSINKI, FINLAND
ECHA.EUROPA.EU
